
目 次
雑科学ホーム
hr-inoueホーム
第1章
磁性粒子表面への樹脂吸着層の形成
1. 緒 言
微粒子分散系の分散安定化に関しては、基本的には粒子の磁性、非磁性によらず同じ考え方が適用できるであろう1)。しかし、磁性粒子を有機溶媒中に分散させる場合、非磁性粒子の分散、あるいは水中への分散と比較して様々な困難がある。まず第一に、磁性に起因する強い引力が粒子間に働くこと2)、第二に、有機溶媒の誘電率が一般に非常に小さいために粒子が帯電せず、静電気的な反発力が働かないことが挙げられよう3)。さらに実用を考えた場合、濃厚な分散液が必要なことが多く4)、ますます凝集しやすい条件が揃うことになる。また、磁性粒子は分散させる前から強固に凝集している場合がほとんどで、分散液作成後の安定化だけでなく、まず初めにこの凝集塊を破壊して一次粒子化することが必要になる。従って磁性粒子の安定な分散液を作製するには、凝集塊の破壊と再凝集の防止という2点を考慮しなければならない。
微粒子の凝集塊を破壊するには機械的な強い剪断力が必要である。そのために、ボールミル、サンドミル、ロールミル、ニーダーなど各種の混合機、混練機が用いられる5)。このような混合、混練の過程を定量的に解釈する試みも行なわれているが6)、非磁性粒子に関してさえも充分に解析されているとは言えない。一方、分散液作成後の再凝集の防止法としては、粒子表面に高分子吸着層を形成して、その立体反発力を利用するということが行なわれている7)。ここでは高分子の種類や吸着層の形態、その作成法などが重要となる。これまでに各種の高分子について分散能力の検討が行なわれており、官能基の効果などが明らかにされている8-11)。しかし、これらの研究で扱われている高分子は熱可塑性のものがほとんどで、固定磁気ディスク媒体等に主に用いられている熱硬化性の高分子についてははあまり検討されていない。また、現実的には、粒子凝集塊の破壊と高分子の吸着は同時に行なわれることが多いが、凝集塊の破壊過程と吸着過程を結びつけて検討した例も少ない12)。
そこで本研究では、磁性粒子表面に熱硬化性樹脂を吸着させて分散液を調整し、分散液の作成条件と吸着樹脂量との関係を調べて、樹脂吸着層の形成過程の解析を試みた13)。磁性粒子としては、磁気記録材料として広く用いられている針状のγ-酸化鉄14)、および最近注目されている板状バリウムフェライト15)を、また熱硬化性樹脂としてはエポキシ樹脂を用いている。さらに、分散液の作成条件を変えて吸着樹脂量の異なる試料を作成し、吸着樹脂が磁性粒子の分散性に与える影響についても検討する。
2. 実験方法
2.1 試 料
磁性粒子としては、針状のγ-酸化鉄および板状のバリウムフェライトを用いた。γ-酸化鉄(γ-Fe2O3)は、スピネル構造を持つマグネタイト(Fe3O4)のFe3+サイトの一部が空孔になった構造をしており、針状にすることにより形状磁気異方性に基づく高い保磁力が得られるため、磁気記録材料として広く用いられている16)。今回用いたのは堺化学製の水熱合成品で17)、ゲーサイト経由で合成したもののような脱水孔を持たないのが特徴である。長軸長は約0.3μm、針状比は約6で、BET比表面積は22m2/g、単位重量当たりの飽和磁気モーメントσsは75emu/g、保磁力Hcは360Oeである。バリウムフェライトはBaFe12O19の組成を持つ六角板状の粒子で、結晶磁気異方性によって板面に垂直な方向に磁化する性質を持っており、磁気記録用として近年注目されている材料である18),19)。合成法としてはガラス結晶化法や共沈法などがあるが、本研究では戸田工業製の水熱合成品を用いた。粒子径は約0.1μm、板状比は1/5、BET比表面積30m2/g、σsは60emu/g、Hcは660Oeである。
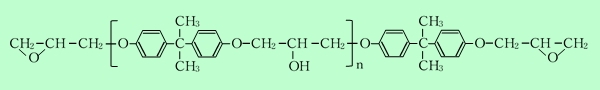
エポキシ樹脂は、油化シェル製のビスフェノール-Aタイプのものを使用した20)。基本骨格を下に示す。数平均分子量は2900および900の2種類である。
溶媒は関東化学製のシクロヘキサノンおよび酢酸2-エトキシエチルを用いた。
2.2 分散液の作製
磁性粒子を分散させるために、まず初めに粒子とエポキシ樹脂とを少量の溶媒と共に高粘度の状態で充分に混練し、その後に希釈するという方法をとった。混練にはニーダーと呼ばれる混練機を使用した21),22)。この混練機は、互いに逆方向に、異なる速度で回転する2枚のブレードにより、ブレードと混練機内壁との間の狭いクリアランスに内容物を畳み込んで強い剪断力を加えるものである。用いた装置は内容量1L 、ブレード回転数30min-1および20min-1、クリアランス1mmで、内容物の状態が変わってブレードにかかる負荷が変化しても、投入電力を自動調節して回転数が一定に保たれるようになっている。
この混練機に700gの磁性粒子と、250gのエポキシ樹脂、および50〜300gの溶媒を入れて混合する。しばらく混合を続けると溶媒が樹脂を膨潤させて全体に行きわたり、混合物全体がひと塊のペースト状になった、いわゆる混練状態となる。溶媒の添加量が少なすぎると混練状態にならないが、50g以上の溶媒を加えれば、遅くとも数時間後には混練状態に到達する。この状態で1〜30時間混練を続ける。混練時に添加される溶媒の量が少ないほど混練物は高粘度になり、強い剪断力がかかると同時に摩擦によって温度も上昇することになる。
混練終了後、ボールミルを用いて溶媒で希釈、分散させる。現実の磁性塗料では、ここでさらに種々のバインダー樹脂や数種類の溶媒が添加されるが、本研究では条件を単純化するために、一種類の溶媒のみで希釈した。24時間程度のボールミル混合により粘度が安定し、均一な分散液が得られるが、完全を期して3日間混合した。最終的な分散液組成は、磁性粒子21wt%、エポキシ樹脂7.5wt%、溶媒71.5wt%である。
2.3 各種測定
作成した磁性粒子分散液を遠心分離機にかけ、上澄み液を採取する。遠心分離は、径20cmのローターを用いて15000min-1で30分間行なった後、その上澄みをさらに同条件で遠心分離するという二段階で行なった。上澄み液をガラス瓶に取り、溶媒を200℃で加熱蒸発させた前後の重量変化から上澄み液の樹脂濃度を求め、吸着樹脂量を算出した。なお、この加熱条件でエポキシ樹脂の重量が変化しないことは確認済みである。
樹脂の分子量分布は、遠心分離後の上澄み液をガラス板上に薄く広げて80℃で減圧乾燥させた後に回収した樹脂をTHFに再度溶解させ、日立製635型ゲル浸透クロマトグラフィー(GPC)を用いて測定した。この程度の乾燥条件ではエポキシ樹脂の分子量分布に変化が起きないことは、あらかじめ確認している。磁性粒子の分散状態は、分散液をアセチルセルロースフィルム上に薄く延ばし、カーボン蒸着の後にフィルムを溶解するレプリカ法により、日立製H-9000透過型電子顕微鏡を用いて観察した。また分散性を評価する別の方法として、分散液をアルミニウム製の円板上にスピンコート法で0.5〜1μmの厚さに塗布し、200℃で加熱硬化した後に、Perthen社製S-5P型ペルトメーターにより薄膜表面の粗さを測定した。分散液の粘度特性は、HAAKE社製ロトビスコRV-12型回転粘度計を用いて測定した。沈降速度の測定は、径2cm、高さ30cmのガラス管に分散液を入れて静置し、2週間にわたって行なった。
3. 結果と考察
3.1 磁性粒子表面への吸着樹脂量
本論文の主題である濃厚分散系について検討する前に、比較として同じ成分の希薄系での吸着現象を概観する。Fig.1に、エポキシ樹脂の針状γ-酸化鉄粒子への吸着等温線を示す23)。この場合の分散液は、種々の濃度のエポキシ樹脂溶液に2wt%のγ-酸化鉄を添加して24時間回転混合して調製している。吸着等温線はLangmuir型で、通常の物理吸着によく見られるように高温にするほど吸着量が減少する24)。Clausius-Clapeyron式を適用して吸着熱を求めると、約50kJ/molという値が得られる。この値は水素結合のエネルギー(20〜30kJ/mol)と比べるとやや大きいが、化学結合エネルギーよりはかなり小さく、この吸着が物理的なものであることが推察される。
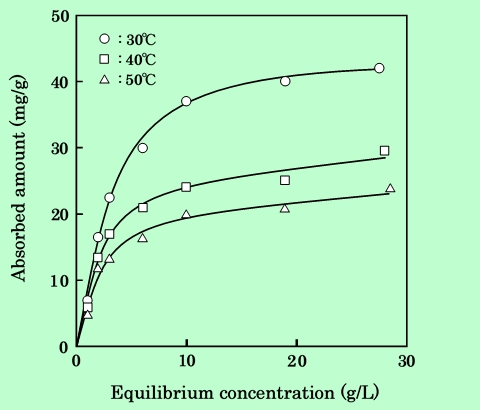
Fig.1 Adsorption Isotherms of epoxy resin on γ-Fe2O3 in cyclohexanone
at various temperatures. Particle concentration is 2 wt.%.
一方濃厚分散系の場合、上記のような希薄系に対して用いた試料作成法では、磁性粒子は充分には分散しない。実験方法の項で述べたような混練過程が不可欠であり、その混練の条件が分散性に強い影響を与えることは容易に予測できる。そこでまず初めに、混練時の温度と吸着樹脂量との関係について検討した。Fig.2は、シクロヘキサノンを溶媒として用いて混練した後に、2種類の溶媒で希釈した場合の吸着樹脂量を示したものである。
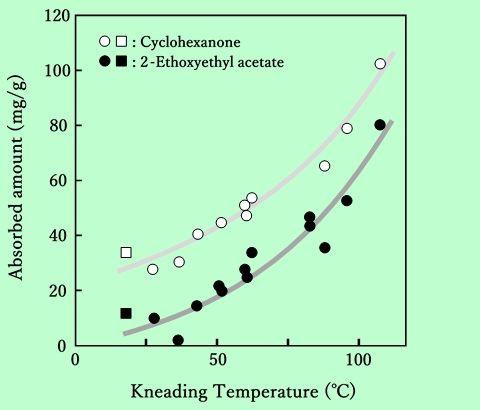
Fig. 2 Dependence of adsorbed amount on kneading temperature
in cyclohexanone and in 2-ethoxyethyl acetate. Square plots
show the adsorbed amounts without kneading process.
図中には、混練過程を経ずに直接ボールミルによって所定濃度の分散液を調製した場合のデータも示している。磁性粒子は針状γ-酸化鉄であるが、板状のバリウムフェライト粒子を用いても傾向は同じである。この図からわかるように、混練温度の上昇に伴って吸着樹脂量は増加する。吸着量の測定はすべて室温で行なっているので、可逆的な物理吸着であるならば吸着量は全て同じになるはずである。しかし現実には吸着量は混練時点での温度によって変わっており、吸着量の絶対値も混練過程を経ない場合よりも増加していることから、この吸着が混練によって生じた不可逆的な化学吸着であることが推察される。赤外線吸収スペクトルの測定から、混練物中のエポキシ樹脂のエポキシ環が開環していることが示されているので25)、おそらく末端エポキシ基が粒子表面の水酸基と
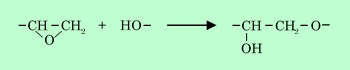
のような反応を起こしているのであろう。
同じ条件で混練した試料でも希釈溶媒が異なると吸着量が変化している。混練条件が同じならば化学吸着量は同じはずであるから、この違いは物理的に吸着している樹脂量の差によると考えられる。このことを確認するために、溶媒を変えて2段階に希釈を行ない吸着量の変化を調べた。Fig.3にその結果を示す。
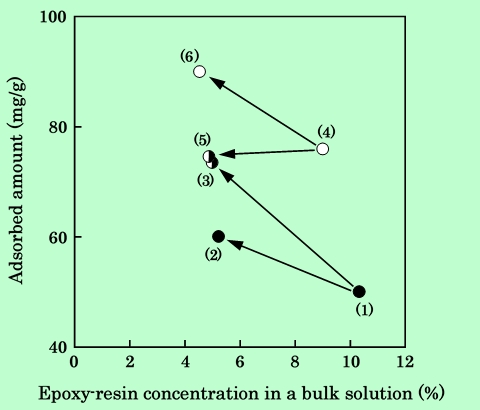
Fig.3 Solvent effect on epoxy-resin adsorbed amount.
Each dispersion was prepared as follow.
Dispersion (1) : Kneaded mixture was dispersed in cyclohexanone.
Dispersion (2) : Dispersion(1) was diluted by cyclohexanone.
Dispersion (3) : Dispersion(1) was diluted by 2-ethoxyethyl acetate.
Dispersion (4) : Kneaded mixture was dispersed in 2-ethoxyethyl acetate.
Dispersion (5) : Dispersion(4) was diluted by cyclohexanone.
Dispersion (6) : Dispersion(4) was diluted by 2-ethoxyethyl acetate.
希釈することによって吸着量が増加しているが、この現象については第2章で詳しく検討する。この図に示すように吸着量の変化は可逆的であり、化学吸着している樹脂とは別の物理吸着樹脂が、溶媒を変えることによって吸脱着していることが確かめられた。この系で、物理吸着した樹脂量と化学吸着した樹脂量の絶対値はそれぞれいくらかということが問題である。酢酸2-エトキシエチルで希釈した場合とシクロヘキサノンで希釈した場合との差、20〜30mg/gは物理吸着分であることは確かである。また、混練過程を経ずにボールミルのみで分散させた場合の吸着量もかなりの部分が物理吸着によるものであろう。以上のことから、測定値のばらつきを考慮に入れると、シクロヘキサノン中に分散させた場合は10〜20mg/g、酢酸2-エトキシエチル中に分散させた場合は30〜40mg/gが物理吸着した樹脂であると考えられる。従って、100℃程度で混練してシクロヘキサノン中に分散させた場合は、吸着樹脂の80%以上は化学吸着していることになる。
溶媒によって物理吸着量が異なる原因は、主として、その溶媒に対する樹脂の溶解性の違いにあると思われる。溶解性が低いほど、液中に留まるとエネルギー的に不利になるので吸着量が増加するはずである。溶解性の目安としてよく用いられるものに、溶解性パラメーター(SP値)がある。SP値で比較すると、シクロヘキサノンと酢酸2-エトキシエチルはいずれも9.9で、エポキシ樹脂は8.5〜13であるから26),27)、この値からは溶解性の違いは見出せない。しかし、より定量的な溶解性の指標である極限粘度の値で比較すると、エポキシ樹脂に関しては、シクロヘキサノンが22.3、酢酸2-エトキシエチルが19.2であり、シクロヘキサノンの方がやや溶解性が高いことになり、吸着量の傾向と一致する。
3.2 樹脂の分子量分布
分子量分布を持つ樹脂を吸着させる場合、吸着相と液相とでその分布に偏りが生じることが多い。そこで、遠心分離後の上澄み液中の樹脂の分子量分布をGPCで測定し、原料樹脂と比較した。Fig.4は、平均分子量2900の樹脂と900の樹脂のGPCチャートである。
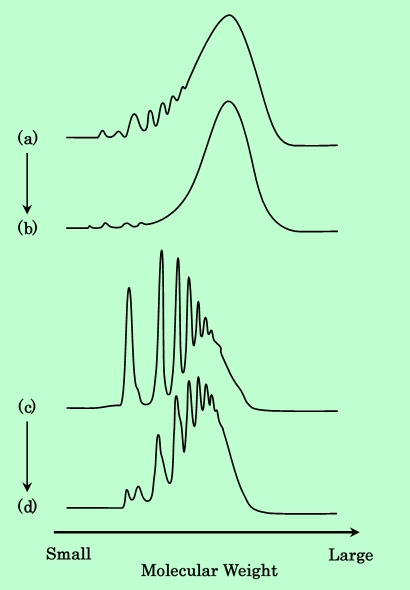
Fig.4 Change of molecular weight distribution of epoxy-resin through
kneading process. Molecular weight distributions were measured by GPC.
(a) : GPC chart of original epoxy-resin (Mn = 2900).
(b) : GPC chart of epoxy-resin in a bulk solution after kneading (Original Mn = 2900).
(c) : GPC chart of original epoxy-resin (Mn = 900).
(d) : GPC chart of epoxy-resin in a bulk solution after kneading (Original Mn = 900).
図の左側が分子量の小さい成分であり、各ピークは単量体および2量体等のオリゴマーに対応している。どちらの樹脂においても吸着後の上澄みでは低分子量の成分が減少していることがわかる。すなわち、低分子量の成分の方が優先的に吸着されていることになる。分子量による吸着性の違いについては第2章でさらに詳しく検討するが、ここではこの性質を利用して、吸着の不可逆性の検証を行なうことにする。
実験の手順をFig.5に、また測定結果をFig.6に示す。
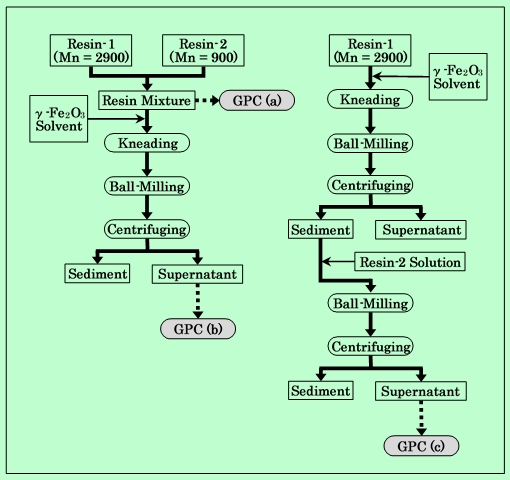
Fig.5 Experiment for confirming irreversibility of adsorption.
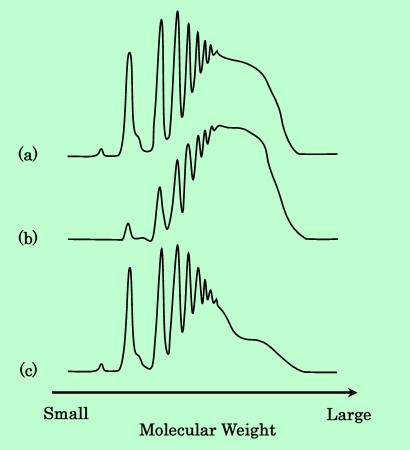
Fig.6 Molecular weight distributions of epoxy-resin prepared
by the process shown in Fig.5.
Chart (a), (b) amd (c) are correspond to the GPC samples (a), (b) and (c).
まず初めに、平均分子量2900の樹脂(樹脂-1とする)と900の樹脂(樹脂-2とする)とを等量混合して、混合樹脂を作成する。これをTHFに溶解してGPC測定を行なう。チャートは当然、Fig.4の2種類の原料樹脂のチャートを重ねたものになる(Fig.6-a)。この混合樹脂を用いて前述の方法で磁性粒子分散液を調製し、遠心分離後、上澄み中の樹脂をGPC測定する。その結果をFig.6-bに示すが、単独の樹脂を用いた場合と同様に低分子量成分の減少が見られる。次に樹脂-1のみで分散液を調製し、遠心分離を行なった後、その上澄みの一部を樹脂-2の溶液で置換してボールミルで3日間混合する。分散液の組成は、2種類の樹脂の配合比も含めて、混合樹脂で調製した場合と同じになるようにする。これを再度遠心分離して上澄み液をGPC測定にかける。測定の結果は、Fig.6-cに示すように、組成が同じであるにもかかわらず、Fig.6-bとは明らかに異なっている。あとから添加した低分子量樹脂は、吸着性が高いにもかかわらず、ほとんど吸着されずに液中に残っており、混練過程で吸着した高分子量樹脂がそのまま吸着されていることがわかる。従って、混練時に起こる樹脂吸着は不可逆的な過程であり、その後の環境の変化によって脱離したり置換されたりすることはほとんどないということが、この分子量分布の測定からも確認された。
3.3 吸着量の混練時間依存性
Fig.2に示したのは全て混練時間4時間のデータである。温度の上昇と供に吸着量が増加することから、この程度の時間ではまだ飽和吸着量には到達していないと考えられる。混練時間の経過と共に吸着量がどの様に変化するかを調べた結果をFig.7に示す。
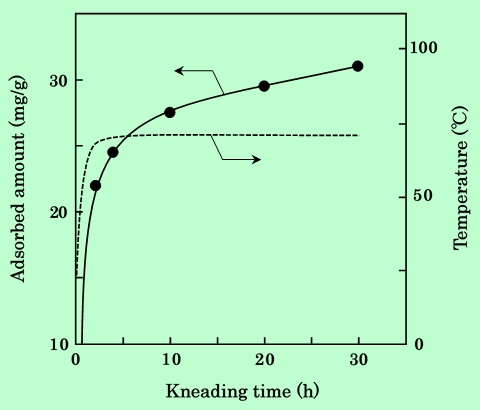
Fig.7 Dependence of epoxy-resin adsorbed amount on kneading time.
Dashed line shows the temperature.
温度の方は混練開始後約2時間でほぼ一定値に到達するが、吸着量は30時間経過した後もまだ飽和する様子は見られない。従って、Fig.2に現れている吸着量の差は、吸着速度の違いを反映したものである、と言える。即ち、高温で混練するほど樹脂の吸着速度が大きくなり、同じ時間で比較すると吸着量が多いことになる。
3.4 樹脂の吸着を促進する要因
実験方法の項で述べたように、混練温度は添加する溶媒の量を変えることで変化させている。この方法では、添加する溶媒量が少ないほど混練物が高粘度になり、強い剪断力が加わるために摩擦による発熱量が多くなって温度が上昇する。そのため、混練条件としては温度のほかに剪断力も同時に変化している。そこで、吸着を促進する要因を明らかにするために、混練時に外部から強制的に加熱または冷却して、温度と剪断力の2つの条件を分離して評価した。加熱すると温度は上昇するが、混練物の粘度は低くなり剪断力が加わらなくなる。一方冷却すると、逆に低温、高剪断力の条件になる。このような種々の条件下で混練を行ない、吸着樹脂量を比較した。剪断力の尺度としては、混練機の消費電力の値が利用できる。今回用いている混練機は負荷によらず一定速度でブレードが回転するようになっているので、混練機が消費する電力が剪断力にほぼ比例するからである。
混練温度と混練機の消費電力とをパラメータとして吸着樹脂量をプロットしたのがFig.8である。まず針状粒子についての結果から検討する(Fig.8-(a))。
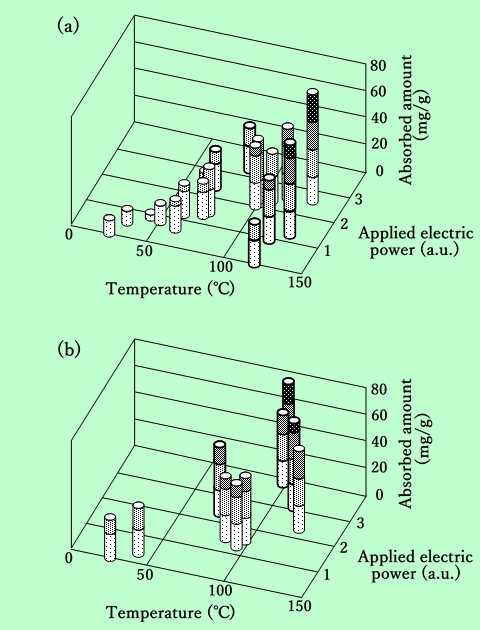
Fig.8 Effect of temperature and shear on epoxy-resin adsorbe
amount. The shear is represented by total electric power
applied to a kneading machine.
(a) : Acicular γ-Fe2O3, (b) : Platelet Ba-ferrite
図の左手前から右奥に向かう対角線上にプロットされているデータはFig.2 と同様で、強制的な加熱、冷却は行なっていない。右奥、即ち高温、高剪断力になるほど吸着樹脂量は増加する。これに対して、図中の太線で示されたプロットは加熱または冷却を行なったもので、左奥は冷却による低温、高剪断力混練、右手前は加熱による高温、低剪断力混練の場合のデータである。左奥のデータを見ると、同じ温度で剪断力が低い場合と比較して、吸着量はほとんど増加していない。つまりこの領域では、吸着量は剪断力ではなく温度で決まることになる。一方、右手前の高温、低剪断力混練では、同じ剪断力の低温混練の場合よりも吸着量は多く、また同じ温度の高剪断力混練の場合よりは吸着量が少なくなっている。従ってこの領域では、温度と剪断力の両方が樹脂吸着に影響を与えていることになる。
この結果は次のように解釈できる。混練過程ではまず機械的な剪断力によって磁性粒子の凝集塊が破壊され、新しい粒子表面が溶媒で膨潤した樹脂と接触する。その時に充分な熱エネルギーが供給されると、樹脂が磁性粒子表面に化学結合する。凝集塊を完全に破壊するには一定以上の剪断力が必要とされるが、それ以上の剪断を加えても無意味であり、吸着速度は温度のみに支配される。しかし剪断力がそれ以下になると、凝集塊の破壊が遅いため、新しい粒子表面の露出過程が律速となる領域に入ってきて、吸着量は両方の因子に支配されるようになる。温度がさらに高い領域では、逆に温度に対して吸着量が飽和する現象が現れることが予測されるが、150℃以上では樹脂の変性が始まるため、これを実験的に確認することはできなかった。
一方、板状粒子に関しては状況がやや異なる。Fig.8-(b)の温度100℃のライン上のプロットからわかるように、剪断力の増加に伴って吸着樹脂量が増加している。このことは、剪断力がまだ不足しており、凝集塊を完全に破壊するまでに至っていないことを示している。即ち、板状粒子の凝集塊を破壊するには、針状粒子よりも強い剪断力が必要である、と言える。
3.5 凝集塊の破壊に要する力
前節で問題となった、磁性粒子凝集塊を破壊するのに要する力が粒子形状によってどう変わるかを、簡単な計算で調べてみた。計算に用いた粒子モデルをFig.9に示す。磁性粒子は正方角柱で近似し、接触している2個の粒子を一定方向にずらせて行くのに要するエネルギーと力を計算する。計算では、図のように粒子を同じ体積の立方体に分割し、各立方体の中心に磁気モーメントを置いて、全モーメント間の磁気的相互作用エネルギーを積算する方法を採った。分割数は、針状粒子が3×3×18、板状粒子が10×10×2である。厳密には、このような粒子間距離が非常に小さい条件下では、分割数をさらに多くするか、または積分を用いない限り正確な値は求められない。また van der Waals力も無視できない大きさになっているはずである。しかし、実際の粒子表面は平坦なわけではないので、計算に必要な粒子間距離を正確に見積もることは不可能で、モデルの精度だけを良くしても無意味である。ここでは粒子形状の比較という意味での粗い近似にとどめることにする。
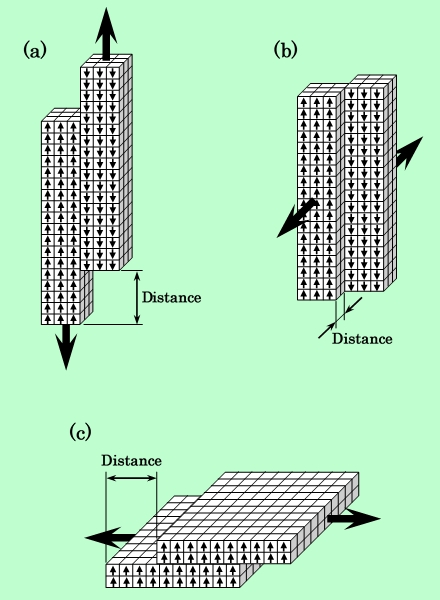
Fig.9 Schematic models used to calculate shear force requirement
to separete two particles.
(a) : Acicular particle sliding in the long axis direction.
(b) : Acicular particle sliding in the short axis direction.
(c) : Platelet particle sliding in the plane direction.
計算結果はFig.10のようになる。図中の(a)、(b)、(c)はFig.9の各モデルに対応しており、粒子の大きさ、および磁化量は実際に用いた試料に合わせてある。2粒子を分離するのに要する力を見ると、板状粒子の方が磁化量が小さいにもかかわらず、針状粒子に比べて1.5〜2倍大きいことがわかる。しかも、2粒子を完全に分離するまで必要な力はほとんど変化せず、分離した直後の反発力も小さい。これらの点が、板状粒子の凝集塊の破壊を困難にする一因となるのであろう。
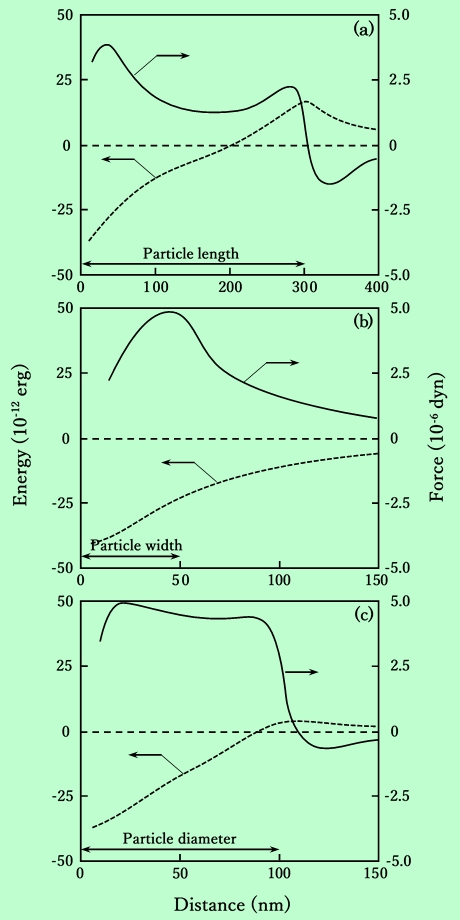
Fig.10 Shear energy and shear force required to separate two particles.
Each figure corresponds to model shown in Fig.9.
3.6 分散性の評価
これまでの検討結果から、混練過程の条件を制御することによって磁性粒子への吸着樹脂量を調節できることがわかった。そこで吸着樹脂量の異なる分散液を種々調製し、その中での磁性粒子の分散性を調べた。分散性を検証する最も直接的な手法は電子顕微鏡による観察であろう。吸着樹脂量の異なる3種類の分散液の透過型電子顕微鏡(TEM)による観察結果をFig.11に示す。

Fig.11 TEM images of magnetic particle dispersions.
Left column : Dispersions just after ball-milling (21 wt.% particles).
Right column : Diluted dispersions (10 wt.%)
Epoxy-resin adsorbed amounts are 44 (a, d), 54 (b, e), and 81 (c, f) mg/g.
左側の列は最終的な分散液(磁性粒子濃度21wt%)の写真で、ボールミルによる混合を3日間行なった直後に撮影したものである。この写真で見る限りは、磁性粒子は一次粒子に近く、かなり良好な分散状態にあると言える。吸着樹脂量の影響はほとんど現れていない。これは実際に凝集が起こっていないのか、あるいはTEM用のレプリカを作成する際に、アプリケーターを用いて試料を薄く延ばす段階で凝集力の弱い粒子塊が破壊されているためかも知れない。そこで、凝集を促進させるために、溶媒を添加して磁性粒子濃度を21wt%から10wt%に希釈し、ボールミルによる混合を1日行なった後に同様にTEM観察を行なった。その結果が図の右側の列に示されている。希釈前の写真と比較して明らかに凝集が進行しており、また吸着樹脂量が少ないほど磁性粒子が激しく凝集していることがわかる。
粒子の分散性は、分散液の粘度特性に大きな影響を与える。一般に、分散性がよくなるほど粘度は上昇すると考えられる28)。粘度特性に関する詳細な議論は第3章で行なうので、ここでは粘性に関するいくつかの代表的な数値を分散性の指標として検討することにする。Fig.12に示したのは、ずり速度14s-1での粘度値である。微粒子の分散液はほとんどの場合ニュートン流体ではないので29)、ずり速度によって粘度の値が異なるのが普通であり、ここで示した値も特定のずり速度におけるみかけの粘度である。
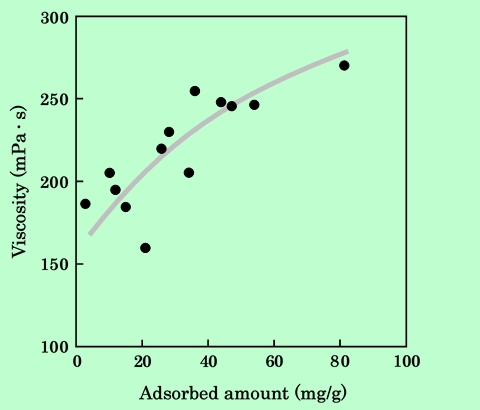
Fig.12 Dependence of viscosity on epoxy-resin adsorbed amount.
Viscosity was measured at shear rate 14s-1.
図からわかるように、吸着樹脂量の増加に伴って粘度は増大する傾向がある。吸着樹脂が磁性粒子の凝集を防止し、分散した粒子間の相互作用が粘度に寄与するようになったためであると考えられる。
前述のように、本研究で扱っている分散液は非ニュートン流体であり、Fig.12に示した値は、みかけ粘度である。このような非ニュートン流体の粘度特性を記述するのに有用な経験式として、次のCassonの式がよく用いられる30)。
τ1/2 =η∞1/2 D1/2 +σy1/2 (1.1)
ここで、τはずり応力、Dはずり速度、η∞はずり速度が無限大のときの粘度に対応する値、σyは降伏値である。ずり応力の平方根をずり速度の平方根に対してプロットすると、流動性を表す特性値としてη∞とσyが得られる。このようにして求めたη∞とσyを吸着樹脂量に対して整理した結果をFig.13とFig.14に示す。
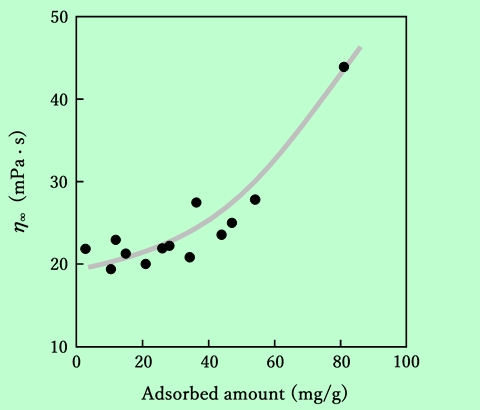
Fig.13 Dependence of viscosity at infinite shear (η∞) on epoxy-resin
adsorbed amount. The value of η∞ was obtained from Casson plot.
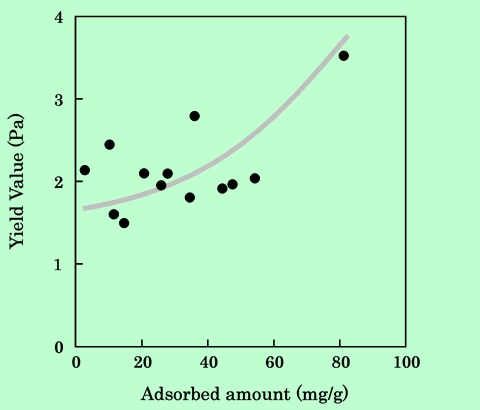
Fig.14 Dependence of Casson's yield value on epoxy-resin adsorbed amount.
σyの方はデータのばらつきが大きいが、どちらもほぼ同じ傾向を示している。従って、Fig.12に現れている粘度の吸着量依存性には、η∞とσyの両方の要素が含まれていることになる。η∞の増大は、粒子の分散性の向上による部分が大きいであろう。一方σyに関しては、単純に分散性の良否を表しているとは考えにくい。第3章でも検討するが、これはおそらく、吸着樹脂を介して粒子間に弱い相互作用が働いて、一種のゲル的な構造が生じているためではないかと推察される。
分散系の安定性を評価する指標として、沈降速度もよく用いられる。Fig.15は、分散液を2週間静置して、その間の平均沈降速度を吸着樹脂量をパラメーターとしてプロットしたものである。
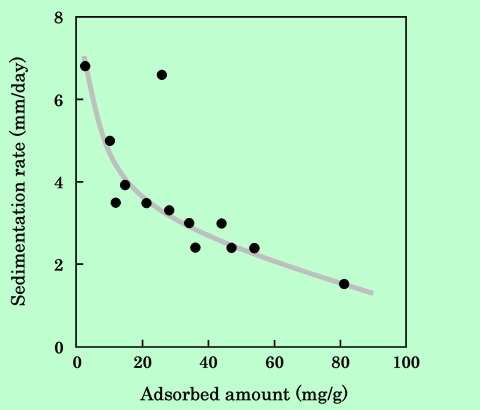
Fig.15 Dependence of sedimentation rate on epoxy-resin adsorbed amount.
吸着樹脂量が多いほど沈降が遅くなり、分散安定性が増していることがわかる。ただし、前述のように吸着樹脂量の増加に伴って分散液の粘度も増加しており、高粘度になれば当然沈降速度は低下すると考えられるので、沈降速度が磁性粒子の凝集状態を直接に表しているとは言いきれない。しかし、仮に粘度の増加という要因を介しているとしても、沈降が遅いほうが分散系の安定度が高いということは事実である。
異なる溶媒を用いたときの分散安定性の違いを、沈降速度の観点から比較したのがFig.16である。
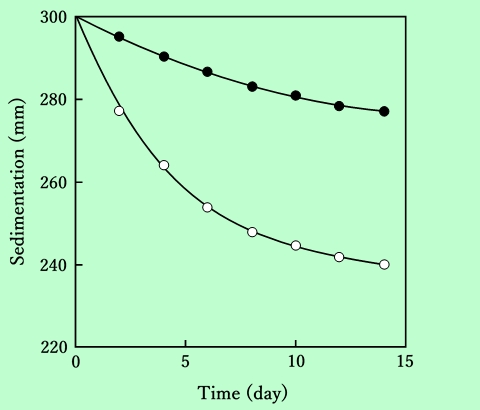
Fig.16 Sedimentation of magnetic particles in cyclohexanone
(black circles) and in 2-ethoxyethyl acetate (white circles).
吸着樹脂量は、どちらの分散液の場合も約50mg/gで、粘度はそれぞれ240mPa・sおよび220mPa・s(共にずり速度14s-1での値)である。この図に示されているように、同じ吸着樹脂量で比較すると、シクロヘキサノン中に分散させた場合よりも酢酸2-エトキシエチル中に分散させた場合の方が、沈降が2倍以上速い。磁性粒子の凝集が速いことと、分散液の粘度が低いこと(これは初期の分散状態の反映とも考えられる)の両方の要因が関与しているのではないかと思われる。このような溶媒効果が現われる機構については、後の章でさらに詳しく検討する。
分散性の評価法として、分散液を基体に塗布して薄膜状にし、その表面粗さを測定する方法も採用した。塗膜が厚いと、膜中で磁性粒子の凝集塊が互いに重なりあって表面は平滑になってしまうが、膜厚が凝集塊の大きさ程度になると、凝集状態が表面粗さに反映されるようになる。Fig.17に測定結果を示す。
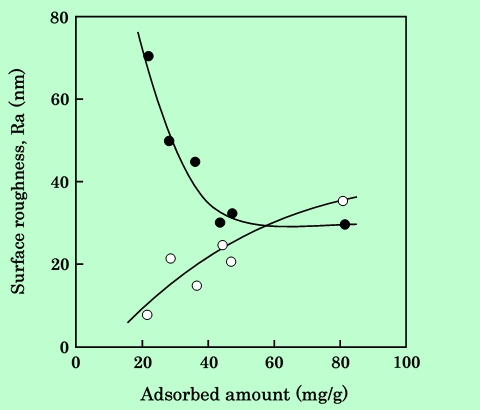
Fig.17 Dependence of surface roughness on epoxy-resin adsorbed amount.
The surface roughness value is divided into long wavelength component
(0.08 mm<λ<0.8 mm, white circles in the figure), and short wavelength
component (λ<0.08 mm, black circles in the figure).
測定装置には、表面粗さの内、波長が一定の値以上のうねり成分をカットする機能があり、この機能を用いて波長が0.08mmよりも長い成分と短い成分とに分けて測定している。両成分は傾向が全く逆で、長波長成分は吸着樹脂量の増加に伴って徐々に大きくなるのに対し、短波長成分は小さくなって行く。短波長成分は、その波長の大きさと吸着量に対する傾向から見て、磁性粒子の凝集塊そのものを捉えていると考えられる。一方、長波長成分は、TEM写真や短波長の粗さに見られるような凝集塊よりも大きな規模の塊が存在することを示している。これは、流動の降伏値からも推定されたように、分散液中にゲル状の構造が生じているためであろう。吸着樹脂量の増加によって、樹脂が磁性粒子の間を橋渡しする形でゲル状構造が形成され、これが塗布の際に分断されて長波長の表面粗さになる、と考えられる。
4. 結 言
磁性粒子として針状γ-酸化鉄粒子および板状バリウムフェライト粒子、分散剤としてエポキシ樹脂を用いて、磁性粒子の有機溶媒中分散液を調製し、その過程で起こる樹脂の吸着現象と、吸着樹脂が粒子の分散性に与える影響について検討した。磁性粒子と樹脂とを少量の溶媒と共に混練することで磁性粒子表面に不可逆的に樹脂吸着層が形成される。この混練過程では、粒子の凝集塊を破壊するための機械的な剪断力と、樹脂を粒子表面に化学結合させるための熱エネルギーの両方が必要であることがわかった。高温、高剪断力で混練することが吸着量を増加させるための条件であり、板状のバリウムフェライト粒子の場合は特に剪断力が重要であることが、実験および簡単なエネルギー計算から示された。また、低分子量の樹脂の方が優先的に吸着するが、初めに高分子量の樹脂を吸着させると、後から低分子量樹脂を添加しても置換は起こらなかった。このことは吸着の不可逆性を示す一つの証拠となる。分散液中では化学吸着した樹脂の他に物理吸着した樹脂も存在する。物理吸着量は溶媒と樹脂との親和性によって変化し、樹脂溶解性の高いシクロヘキサノン中では、溶解性の劣る酢酸2-エトキシエチル中よりも、物理吸着量が少なかった。
吸着樹脂量が増加すると磁性粒子の分散性が向上し、分散液粘度の増加、沈降速度の低下が起こることが示された。粒子の分散性の向上は、透過型電子顕微鏡による観察や、分散液を薄膜状に塗布したときの塗膜表面粗さの測定からも確認できた。また、流動の降伏値の測定や塗膜表面粗さの長波長成分の測定から、単純な粒子凝集塊とは別のゲル状構造が、吸着樹脂量の増加によって分散液中に形成されることが推察された。
文 献
1)例えば、 原崎勇次, "コーティングの基礎科学", p217, 槙書店 (1977)
2)A.M.Homola and M.R.Lorenz, IEEE Trans. Magn., MAG-22, 716 (1986)
3)E.J.W.Verwey and J.Th.G.Overbeek, "Theory of the stability of Lyophobic Colloids", Elsevier, Amsterdam (1948)
4)赤城元男, "分散・凝集の解明と応用技術", p505, 北原文雄 編, テクノシステム (1992)
5)釣谷泰一, "サスペンションを中心とした分散技術と工業的応用の実際 総合資料集", p279, 分散技術研究会 編, 経営開発センター出版部 (1978)
6)佐藤宗武, 宮南 啓, 粉体工学会誌, 23, 458 (1986)
7)D.H.Napper, J. Colloid Interface Sci., 58, 390 (1977)
8)渡谷誠治, 角谷賢二, 端山文忠, 直野博光, 松本恒隆, 高分子論文集, 35, 555 (1978)
9)角谷賢二, 中前勝彦, 渡谷誠治, 端山文忠, 松本恒隆, 高分子論文集, 37, 49 (1980)
10)角谷賢二, 秦井俊明, 中前勝彦, 松本恒隆, 高分子論文集, 38, 139 (1981)
11)中前勝彦, 表面, 25, 401 (1987)
12)S.Dasgupta, J. Colloid Interface Sci., 124, 22 (1988)
13)H.Inoue, H.Fukke, M.Akagi and M.Katsumoto, J. Magn. Magn. Mater., 118, 263 (1993)
14)富永匡昭, "磁気記録媒体総合資料集", p31, 松本光功 編, 総合電子出版社 (1985)
15)K.Kubo, T.Ido and H.Yokoyama, IEEE Trans. Magn., MAG-18, 1122 (1982)
16)近角聰信, "強磁性体の物理(下)", p10, 裳華房 (1984)
17)松本清治, 色材, 57, 602 (1984)
18)R.G.Simmons, IEEE Trans. Magn., 26, 93 (1990)
19)N.Kodama, H.Inoue, G.Spratt, Y.Uesaka and M.Katsumoto, J. Appl. Phys., 69, 4490 (1991)
20)岡山清明, "新エポキシ樹脂", p18, 垣内 弘 編, 昭晃堂 (1985)
21)斎藤勝義, セラミックス, 22, 385 (1987)
22)公開特許公報 昭63-4422
23)河合武司, 尾野文治, 今野紀二朗, 井上 均, 勝本正之, 日本化学会第61春季年会 講演予稿集 I, 165 (1991)
24)高橋 彰, 日本接着協会誌, 22, 645 (1986)
25)鍛示和利, 小林憲雄, 栃木憲治, 日本化学会第58春季年会 発表予稿集 I, 771 (1989)
26)C.H.Hansen, Ind. Eng. Chem. Prod. Res. Dev., 8, 2 (1969)
27)"Polymer Handbook", VII/519, J.Brandrup and E.H.Immergut, Wiley Interscience, New York (1989)
28)S.Dasgupta, IEEE Trans. Magn., MAG-20, 7 (1984)
29)松本孝芳, 表面, 25, 590 (1987)
30)N.Casson, "Rheology of Dispersion Systems", p84, C.C.Mill, Pergamon Press, London (1959)
目 次
雑科学ホーム
hr-inoueホーム