
雑科学ホーム
hr-inoueホーム
● 化学結合の話 ●
化学結合は「量子」の世界
世の中の全ての物質は原子が様々につながってできている、ということは、今さら言うまでもありません。その原子どうしの結合には電子が関わっており、電子の関わり方によって、イオン結合や共有結合、金属結合などがあることも、中学や高校で習っているでしょう。ここでは、このような化学結合の一般的な話ではなくて、もう少し突っ込んだ、量子化学の世界を紹介してみようと思います。
我々が生活するマクロな世界では、「粒子」と「波」とは分けて考えることができます。例えば、ここにボールがあるとしましょう。このボールは1個2個と数えることができます。転がすと(余分な力が加わらない限りは)真っ直ぐ進み、狭い隙間を通り抜けてもそのまま直進するのみです。これらは「粒子」の性質、と言うことができます。これに対して、海の波や
音波などはどうでしょうか。これらは、ある程度の広がりを持っていて、「何個」と数えることはできません。また、真っ直ぐ進むだけではなく、障害物の隙間や角のところから陰の部分に回り込んだり(回折)、複数の波が重なり合って縞模様を作ったり(干渉)もします。これらは「波」の特徴的な性質で、先の「粒子」とは全く違います。どんな変化球を投げようと、ボールが回折や干渉を起こしたりすることはないのです。
ところが、原子や電子のようなミクロ(ナノ?)な世界では、同じものが「粒子」の性質と「波」の性質の両方を示します。これは、多数の水や空気の分子が連動して海の波や音波を作るのとは全く別モノで、それ自体が「粒子」でもあり「波」でもあるのです。例えば、普通は波と考えられている「光」でも、一定のエネルギーを持った塊、つまり粒子として振舞う場合がありますし、普通は粒子と考えられている「電子」でも、回折や干渉などの、波でしか見られないはずの性質を見せます。マクロな世界の常識ではちょっと考えられないような状況ですが、これがまさしく「ミクロな世界の現実」であり、その振る舞いがあまりに微小なので、マクロな世界では全く見えなくなっているだけなのです。そしてこの微小な世界の状態を扱うのに必要になるのが「
量子論」です。
となると、化学結合は正に原子や電子の世界なのですから、化学結合を考えるにはどうしても「
量子論」の扱いを取り入れなければなりません。単純に「2個の原子が電子を1個ずつ出し合って共有結合を作る」という解釈で済ますわけには行かないのです。「量子の世界」に関する一般的な話は
量子論の話に詳しく書いていますので、そちらを参照してもらうとして、本稿では、この「化学結合」を量子論で見るとどのようになるか、ということを、ザッと眺めてみることにします。と言っても、かなり複雑な話なので、少々長くなりますが・・・・。
電子の状態を表す便利な道具 ― 波動関数 ―
化学結合の主役は言うまでもなく電子ですから、その電子がどのような状態になっているかを知ることが重要です。バラバラの原子の時に電子の状態がどうなっていて、結合した時はどうなっているかがわかれば、これでもう化学結合の全体像が見えたことになるでしょう。それでは、その電子の状態はどのようにして表したらよいのでしょうか。
太陽の周りを回る惑星のように、電子という粒が原子核の周りを一定の軌道で回っているのならば話は簡単です。軌道の形や径がどれくらいかを調べれば済むでしょう。ところが実際には電子は波のような性質を持っていますから、そう簡単には行きません。波の状態をうまく表してやる必要があります。ここで登場するのが、波の状態を表す便利な道具、「波動関数」です。
波動関数というのは、何も量子論に限った話ではありません。例えばロープの端を持って上下に振った時にできるような普通の波に対しても使われます。このような波には、「周期的に変化する」、「値が大きくなったり小さくなったりする度合い(要するに微分値)も、元の波と同じように変化する」、「位置による変化と、時間による変化が同じような形になる」などの特徴があります。これらの特徴を数式で表したのが「波動方程式」と呼ばれるもので、これを解くと、波の状態を表す「波動関数」が出て来ます。何やらややこしい感じがしますが、多くの人は、波を表す三角関数を使った式を見たことがあるでしょう。高校の物理や数学の教科書にも、普通に出て来ますね。実はこれが、波動関数そのものなのです。普通の波の場合には、波動方程式などは意識しないで直接に波動関数を考えた方が話が簡単です(教科書などでも、波動方程式は考えないで、いきなり波の式が出て来る場合がほとんどだと思います)。必要ならば、波動関数が答になるような波動方程式を後から考えてやればいいのです。
ところが電子の場合には、普通の波と違って、波動関数がどんな形をしているのかサッパリ見当が付きません。こうなると、まず波動方程式を考えて、それを解くことで波動関数を導くしかありません。それではどんな波動方程式を作ればよいのでしょうか。普通の波と同じでいいのでしょうか・・・・。実は、普通の波と同じ式ではダメです。電子には波としての性質の他に粒子としての性質もありますから、これも盛り込んでやらなければならないのです。詳しい説明は省略しますが、普通の波動方程式に修正を加えて、このような粒子の性質と波の性質を両方盛り込んだ形にしたのが、例の有名なシュレーディンガーの波動方程式です。そしてこの方程式を解くことで、波動関数の形とそのエネルギーを知ることができるのです。
ところで、「電子の状態を表す波動関数」というのは、いったい何を意味しているのでしょうか。実は、シュレーディンガーの波動方程式が出された初期の頃は、何やら答は出て来たものの、それが何を意味しているかはわからなかったようです。後になって、この波動関数を2乗した値が、その場所で電子が見つかる確率を表す、ということがわかり、ようやく波動関数に意味付けがされました。電子は空間にボワッと広がっている、と言うよりも、実際には、「どこにでもいる可能性があり、捕まえることで初めて居場所が特定される」、という性質のものです。波動関数(の2乗)は、空間のどこかに電子を捕まえに行った時に、そこでうまく電子が捕まる確率を表したものだったのです。その意味で、波動関数は空間で電子が飛び回る場所、つまり電子の「軌道」を表している、と言うことができます。
実際の波動関数はどんな形? ― 水素原子の場合 ―
シュレーディンガーの波動方程式を解くと波動関数が求まる、とは言うものの、この方程式を厳密に解くことができるのは、実は原子核が1個、電子が1個の場合、つまり水素原子だけです(別の原子でも、1個を残して他の電子を全部取り去ってイオンにしてしまえば厳密計算できます)。その解き方については専門書を見ていただくとして、ここでは結果だけを見て行きます。
まず、答は一つだけではなくて、たくさん出て来ます。そしてその波動関数(軌道)には、特徴的な3つの整数が含まれています。これらの値がピッタリ整数になることで、波が空間内にうまく収まって安定な状態(定常状態)になるのです。定常状態というのは、大雑把に言えば、一周回った時にピッタリ収まるサイズの波になっているということで、何周回っても毎回同じ位置に山や谷が来る状態です。これが1.5とか4.3とかの中途半端な値になると、回るたびに位置がズレて、波が安定に存在できなくなってしまうのです。
ちょっと専門的になりますが、これらの整数はそれぞれ、「主量子数」「方位量子数」「磁気量子数」と呼ばれます。主量子数は正の整数ならばどんな値でもとれますが、他の2つには制限が付いて、可能な組合わせは表Iのようになります(この制限を守らないと、式中の√の中がマイナスになってしまうのです)。
表I 量子数の組み合わせ
| 主量子数 |
方位量子数 |
磁気量子数 |
軌道の名前 |
電子殻 |
| 1 |
0 |
0 |
1s |
K殻 |
| 2 |
0 |
0 |
2s |
L殻 |
| 1 |
-1, 0, 1 |
2p |
| 3 |
0 |
0 |
3s |
M殻 |
| 1 |
-1, 0, 1 |
3p |
| 2 |
-2, -1, 0, 1, 2 |
3d |
| 4 |
0 |
0 |
4s |
N殻 |
| 1 |
-1, 0, 1 |
4p |
| 2 |
-2, -1, 0, 1, 2 |
4d |
| 3 |
-3, -2, -1, 0, 1, 2, 3 |
4f |
・
・
・ |
主量子数は、軌道のエネルギーを表しています(主量子数が同じならばエネルギーは同じです)。後で出て来ますが、主量子数が大きいほど電子は外に広がって行きますので、これを原子核を囲む殻に見立てて、内側から順にK殻、L殻、M殻・・・・、と呼んだりします(電子殻に名前を付ける時に、もっと内側にも新しい殻が見つかることを想定してA〜Jを空けておいたらしいのですが、結局無駄に終わりました)。これに対して方位量子数や磁気量子数は、空間での軌道の広がり方やその方向を表します。方位量子数が0,1,2,3・・・・の軌道をそれぞれ、s軌道、p軌道、d軌道、f軌道・・・・と呼びますが、この名前は、それぞれの軌道が関わる発光現象に因んで付けられたもので、sharp、principal、diffuse、fundamentalの頭文字です(その後は、めんどうになったのか、g、h、i・・・・とアルファベット順になっています)。
それでは、具体的な波動関数(軌道)の形を見てみましょう。波動関数自体は空間に大きく広がっていますから、はっきりした丸や四角の形が外から見えるわけではありません。そこで普通は、同じ値になる部分を結んで、その形で表します。山の高さを表す等高線(この場合は等高"面")のようなものですね。図1(a)〜(c)には、このような等高面で表したs、p、d軌道の概形を示しています(f軌道から先は省略)。赤色の部分と青色の部分は符号が逆で、波の山の位置と谷の位置がちょうど反対になっている、と思ってください。この色(符号)の違いは、後で軌道が重なって化学結合を作る時に重要になります。また、図の右側には、波動関数の断面も示しておきました。色が濃いほど値が大きい(つまり電子の存在確率が高い)ことを表しています。
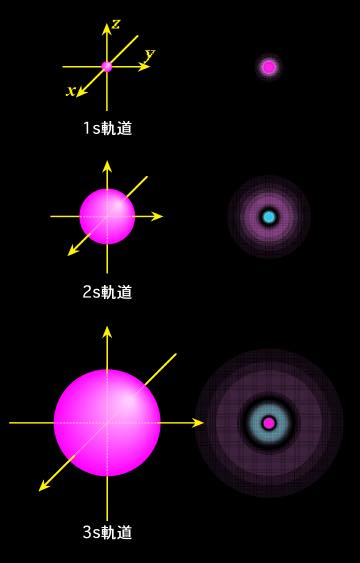
図1(a) s軌道の概形と断面
図1(a)に示したように、s軌道は丸く、球状に広がっています。主量子数が大きくなるほど大きく広がるようになりますが、違いは大きさだけではありません。断面図からわかるように、2s軌道や3s軌道には、途中に波動関数の値がゼロ、つまり電子が存在しない部分ができます。これを「節」あるいは「節面」と言って、主量子数が大きくなるほどその数は増えて行きます。皮と身の間に隙間のできたミカンのような感じでしょうか。
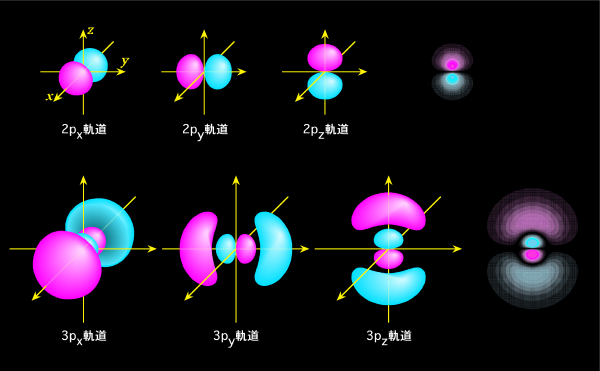
図1(b) p軌道の概形と断面
続いて、図1(b)はp軌道です。p軌道には、表Iに示したように、磁気量子数が異なる3種類があります。このうち、磁気量子数が0の軌道(pz軌道)は問題ないのですが、他の2つは、式の中に山や谷の位置(位相)がズレた波が含まれていて、そのままでは図に描くことができません(数学的には虚数を含んだ形になっています)。そこで、この2つを足したり引いたりして位相が違う部分(虚数の部分)をうまく消し、px軌道とpy軌道という2つの軌道に組み替えてしまいます。こんなことをして大丈夫か、と思うかもしれませんが、出て来た答を足したり引いたりしてできる関数もちゃんと答になる、というのは波動方程式の特徴の一つです。空間をいくつかの波動関数に分ける時の組合わせ方が変わるだけで、元々□+△、□−△という組み合わせだったのを、足し算して□+□、引き算して△+△という形に組み替えたに過ぎません。あるいは、ちょっと乱暴な例えですが、正方形を分割して表す時に、4個の小さい正方形の集まりとして表しても、4個の直角二等辺三角形の集まりとして表してもかまわないのと似たようなもの、と思ってもらえばよいでしょう。注意が必要なのは、波動関数の性質上、組み換えに使った元の軌道の数と、新しくできる軌道の数は必ず同じになる、ということです。元の軌道が2つなら、組み替えて新しくできる軌道も2つなのです
p軌道は、よく亜鈴(ダンベル)型と言われるように、真ん中に電子の存在しない節面があり、その両側で符号が逆になります。またs軌道と同じように、主量子数が大きくなると、さらに新たな節面が途中にできて来て、マトリョーシカ(ロシアの人形)のような入れ子構造になります。
最後にd軌道ですが、d軌道には磁気量子数が5つありますから、全部で5種類になります。この中で、磁気量子数が0のdz2軌道はそのままでよいのですが、他の4つは位相の違う波(虚数の部分)を含みますので、p軌道と同じように組み替えをやります。まず、磁気量子数が-2と+2の2つを組合わせて、dx2-y2軌道とdxy軌道を作ります。さらに磁気量子数が-1と+1の2つを組合わせてdyz軌道とdzx軌道ができます。これらを示したのが図1(c)です。
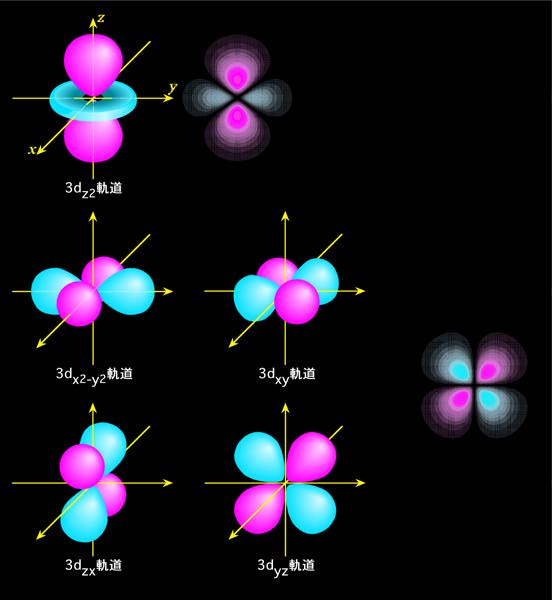
図1(c) d軌道の概形と断面
d軌道の場合、最低でも2つの節面があることがわかります。dz2軌道だけは節面が円錐形ですが、他の4つの軌道では節面は2つの直交する平面になります。また図には示していませんが、主量子数が増えれば、やはり入れ子構造になって節面が増えて行きます。
さて、ここでもう一度図1(a)のs軌道の断面図を見てみましょう。波動関数の値が一番大きいのは中心部分、つまり原子核のあるところですから、電子は原子核にくっ付いている確率が最も高いのでしょうか。実は、ここにはちょっとしたカラクリがあります。同じ体積当たりで比較すれば、確かに中心部が最も値が大きくなります。しかし、中心から一定の距離に電子がいる確率、ということになると、中心から離れるほど体積が大きくなりますから、トータルの確率は、中心部が最大というわけではなくなるのです。これを実際に計算した結果が図2です。
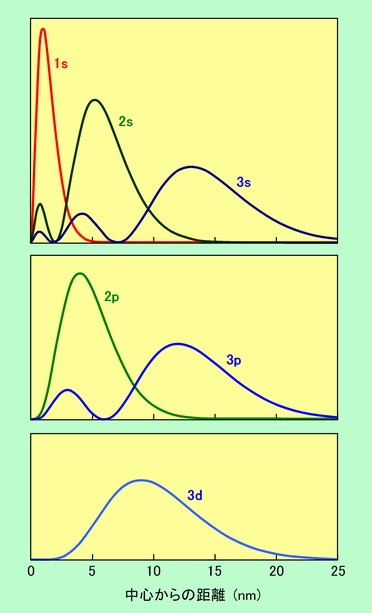
図2 中心から一定の距離に電子がいる確率(動径密度分布)
これを動径密度分布と言って、この図を見れば、電子が原子の中心からどれくらいの距離にいる可能性が高いかがわかります。例えば水素原子の1s軌道では、中心から0.05nm附近が最も確率が高くなっています。また他の軌道でも、図1の波動関数の形に応じて、何箇所かの極大点が見られます。このように電子の分布確率は案外偏っていて、特定の距離の近辺にいる可能性がけっこう高いのです。こうして見ると、太陽の周りを回る惑星のような古典的なモデルも、あながち的外れでもないように思えますね。
水素原子の話の最後に、軌道のエネルギーについても見ておきましょう。波動方程式を解いて出て来る軌道のエネルギーは図3のようになります(原子核から無限に遠く離れた場所のエネルギーをゼロとしています)。
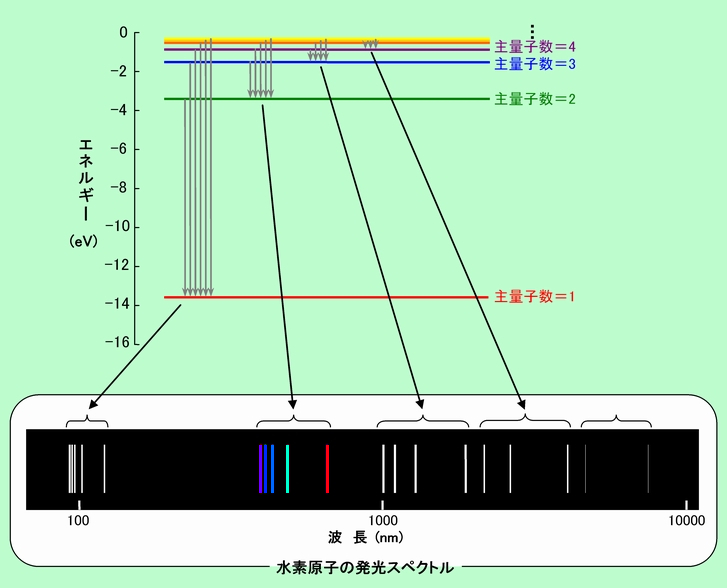
図3 水素原子の軌道のエネルギー
主量子数が飛び飛びの値ですから、エネルギーも連続ではなく、飛び飛びになっています。これが
量子論の大きな特徴の一つですね。各軌道のエネルギーはマイナスですから、そこに電子が収まると、その分だけエネルギーが小さくなる、つまり安定になります。ダントツに安定なのが主量子数=1の軌道ですから、水素原子の電子は、普通はこの
1s軌道に入ることになります。
ただし、光を当てたり温度をものすごく高くしたりすると、
1s軌道にあった電子が高いエネルギーの軌道に飛び上がる場合があります。高いエネルギーに上がった電子は不安定ですので、簡単に下の軌道に落ちるのですが(図中の灰色の矢印)、この時に、そのエネルギー差に相当する光を出します。これが水素原子の
発光現象です。軌道の組合わせによって様々な波長の光が出ますから、これを観測すると、図3下に描いたようなスペクトルが得られます。水素原子の発光スペクトルがこのような飛び飛びの線になることは古くから知られていましたが、以前はその理由を全く説明することができませんでした。量子論の登場によって初めて理論的に解釈することができるようになったのです。
-------------------------------------------------------------------------
既にご存知の方も多いと思いますが、水素原子の発光スペクトルには、落ちる先の軌道ごとに名前が付いています。主量子数=1の軌道に落ちる場合の一群のスペクトルが「ライマン系列」、主量子数=2の軌道に落ちるのが「バルマー系列」、主量子数=3の軌道に落ちるのが「パッシェン系列」、主量子数=4の軌道に落ちるのが「ブラケット系列」です。ライマン系列はエネルギーが大きいので紫外線の領域、バルマー系列はややエネルギーが小さくなって、主に可視光線の領域、その他は赤外線の領域になります。
-------------------------------------------------------------------------
電子が2個以上の場合(水素以外の原子)の考え方
シュレーディンガーの波動方程式を厳密に解くことができるのは原子核と電子が1個ずつの場合だけ、と書きました。それでは電子をたくさん持つ普通の原子はどうすればいいのか、と言うと、厳密に計算するのはあきらめて、何とかして、おおよその答を求めるしかありません。これをカッコいい(?)言葉で、「近似」と言います。
近似の方法にはいろいろありますが、よく使われるのは、とりあえず適当な波動関数の形を仮決めしておいて(この中には値が決まっていないパラメーターがいくつか含まれています)、エネルギーが最も小さくなるようにパラメーターを選ぶ、というものです。この場合、当然ながら、初めにどんな波動関数を設定するかが重要になります。
最も単純なのは、全ての電子に対して水素原子の波動関数がそのまま当てはまると考えて、水素の波動関数の掛け合わせで表す方法です(電子が10個ならば10個の波動関数を掛け合わせます)。この時、まるっきり元の波動関数のまま、というわけには行きませんから(それだと改めて計算する必要もありませんから)、他の電子のマイナス電荷の影響で原子核のプラス電荷が一部隠されて小さくなる、という条件をつけて、より現実的な状況にしてやります。とは言うものの、全ての電子に対して水素原子の波動関数をそのまま使うということは、他の電子があろうがなかろうがお構いなしに、それぞれの電子が水素原子としての独自路線を貫く、ということを意味するわけですから、かなり乱暴な近似であることは確かです。そこで、水素の波動関数にこだわらないで、とにかく電子1個の波動関数の掛け合わせで全体の波動関数が表される、という設定だけでスタートする方法が考えられました。
水素原子の波動関数は他の電子がないという条件で求められたものでした。この制限を外すのですから、他の電子の影響、つまり電子どうしの反発も含めることになり、近似の精度は上がります。しかし、波動関数を一発で計算して求めることができないので、まず適当な波動関数を仮定して計算し、そこから出て来た波動関数(普通は初めに仮定した波動関数とは食い違います)を使って再度計算し、さらにそこから出て来た波動関数で計算し・・・・という風に、食い違いがなくなるまで延々と計算を繰り返さなければなりません。これを手で計算するのは大変ですが、コンピューターにとっては単純作業の繰り返しは最も得意とするところですから、今では十分な精度の計算が可能になっています。
ここまでやればほぼ完璧、と言いたいところですが、まだ問題がありました。電子にはもうひとつ「スピン」という性質があって、これも考えてやらなければならないのです。詳しい話は省略しますが、スピンも含めて考えると、全体の波動関数を表すのに、電子1個の波動関数の単純な掛け合わせだけでは具合が悪く、ちょっと修正が必要になります。ここまで来てようやく、いろいろな実験事実ともよく合う、満足な結果が得られるのです。
計算で求められた波動関数は、同じシュレーディンガーの波動方程式から出発しているのですから、水素原子の場合と似たような形になり、1sとか2pとか3dなどの軌道が出て来ます。ただし、原子番号の大きい原子では原子核の電荷が大きくなるので、エネルギーの値は水素原子とはかなり違って来ます。図4に、いろいろな原子の電子のエネルギーを示しました。図には、各軌道に入っている電子も●で示しています。同じ波動関数の軌道には、スピンを逆にした2個の電子しか入れない、という重要な性質がありますから(詳細は省略)、各軌道に入る電子は2個までです。また、電子はエネルギーの低い軌道から順に詰まって行きますので、図のような電子の配置になるのです。
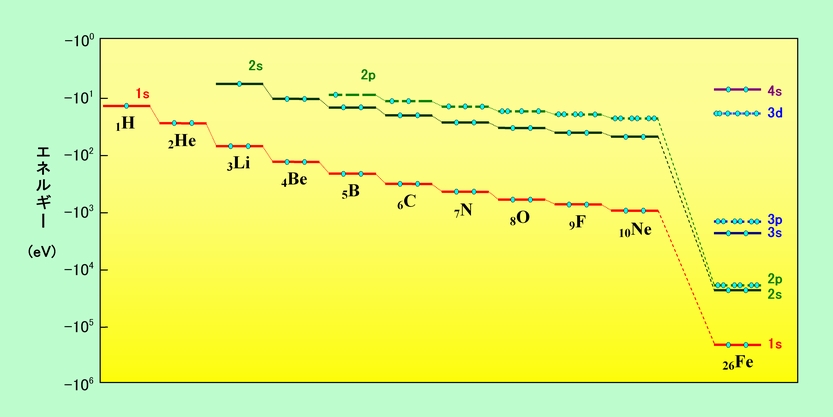
図4 いろいろな原子の電子のエネルギー
原子番号が大きくなるにつれて原子核が大きなプラス電荷を持つようになりますから、それだけ電子を強く引きつけます。そのため、同じ軌道で比べれば、原子番号が大きくなるほど電子は中心に近付き、エネルギーは低く(マイナス方向に大きく)なります(結果的に、電子の数が増えても、全体の電子が広がる範囲、言い換えれば原子の大きさはそれほど変わらない、ということになります)。また、水素の時は主量子数が同じならばエネルギーは同じでしたが、それぞれの軌道に電子が入って来ると、s軌道よりp軌道、p軌道よりd軌道の方がエネルギーが高い状態になります。このようになる理由は、図2の動径密度分布を見ればわかります。例えば2s軌道と2p軌道を比べると、2sの方が内側に一山あり、より原子核の近くに電子がいる可能性が高くなっています。そのため原子核のプラス電荷が2s電子のマイナス電荷に隠されて、2p電子を引き付ける力が弱められ、その分2p電子のエネルギーが高くなるのです。
化学結合にも近似が必要
これまで1個の原子の電子状態(原子軌道)について長々と説明して来ましたが、ここからはいよいよ本題の原子どうしの結び付き、化学結合の話に入ります。1個の原子ではなくて、複数の原子が関わっている状態でシュレーディンガーの波動方程式を解くのです。ところが、ここで大きな問題が発生します。先に書いたように、電子が2個になるともう波動方程式を厳密に解くことはできませんでした。化学結合の場合には最低でも原子核が2個、電子が2個ありますから、厳密な計算は到底不可能なのです。
こういう場合は、やはり近似で何とか格好をつけるしかありません。その近似には、大きく分けて2つの方法があります。一つは「原子価結合法(Valence Bond Method = VB法)」、もう一つは「分子軌道法(Molecular Orbital Method = MO法)」です。どちらもシュレーディンガーの波動方程式を使い、計算をして行く過程もよく似てはいますが、基本となる考え方は全く違います。原子価結合法は、各原子が結合するための「手」を伸ばして隣どうしがつながるイメージですが、分子軌道法は、初めから分子全体に広がる軌道を考えるのです。ブロックを組み上げて行く時に、それぞれのパーツの決まった場所に接着剤を塗って隣どうしをくっ付けて行くのが原子価結合法。目的の形に積上げた後で隙間に接着剤を流し込んで全体を固めるのが分子軌道法、と言えるでしょうか。
電子という「手」を出し合って結合する ― 原子価結合法 ―
まず、原子価結合法を見て行きましょう。この方法では、2個の原子がそれぞれ電子を持っていて、その電子が互いの間で入れ替わったり元に戻ったりして共有されることで結合ができると考えます。電子の交換は隣り合った原子でないとできませんから、結合は隣の原子との間にだけ作られます。この考え方は、原子が価電子という「手」を持っていて、その手で隣の原子と共有結合を作るという(中学や高校で習う)従来の考え方とよく合います。化学構造式や分子模型などのイメージにもピッタリ当てはまりますから、感覚的に理解しやすいと言えるでしょう。これを水素分子について模式的に表現したのが図5です。電子がいる範囲は本当はボワッと広がっているのですが、それでは描きにくいので、ここでは昔ながらの円形の軌道で表しています。
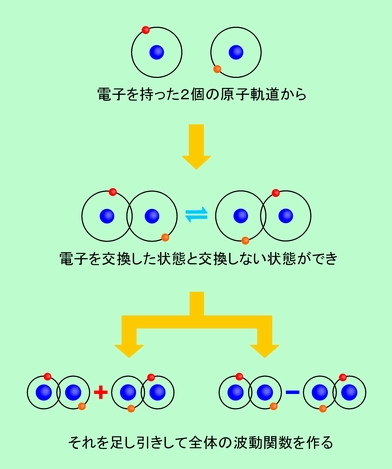
図5 原子価結合法では、電子の交換で結合を作る
実際に計算する時には、まず各原子が結合に関わる電子を1個持った波動関数(軌道)を考えます。これまでに出て来た「原子軌道」ですね。原子が接近して来ると、それぞれの原子軌道が重なるようになり、電子は元々の所属を離れて、互いに入れ替わった状態になるケースも出て来るでしょう。これを、化学結合ができた状態と考えます。ここで「結合状態の波動関数」を作りたいのですが、結合状態では「電子が元の所属のままである状態」と「電子が入れ替わった状態」が混在していますから、この2つを足し合わせて全体の波動関数を作ることにします。結合状態の本当の波動関数は厳密には求めることができないので、「電子の交換」に関する2つの状態の組み合わせで近似するのです。
これで、曲がりなりにも結合状態の波動関数を作ることができたわけですが、2つの状態をどういう比率で組合わせるか、ということはまだ決まっていません。そこで、この未確定の要素を含んだままの波動関数をシュレーディンガーの波動方程式に放り込んで、エネルギーが最小になる比率を探します。自然現象は最もエネルギーが小さい状態に落ち着くものですから、このようにすることで、最もありそうな波動関数の形や、その時のエネルギーを具体的に決めることができるのです。
水素分子について実際に計算してみると、2つの状態の組み合わせ方として、単純に両者を足し算した形の波動関数と、一方の符号を逆にして足し算した(要するに引き算した)形の波動関数がセットになって出て来ます。そしてそれぞれの波動関数のエネルギーを、2個の原子間の距離を変えて調べると、図6のようになります。
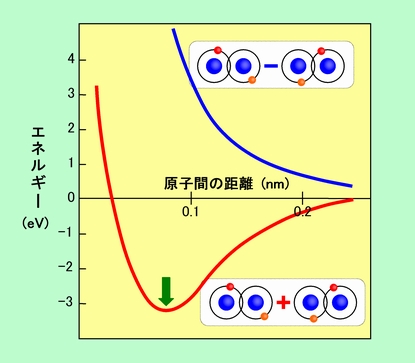
図6 原子価結合法で計算した水素分子のエネルギー
引き算した方は、2個の原子がずっと遠くに離れた状態(つまり結合しないでバラバラになっている状態)よりもエネルギーが高くて不安定です。わざわざこのような不安定な状態になることは、普通はありません。これに対して足し算した形の方は、エネルギーが大きく下がりますから、水素分子としてはこちらの状態になるはずです。また、エネルギーは特定の距離のところ(図6の緑の矢印)で最も小さくなりますから、ちょうど原子と原子がこの距離に来たところで安定する、ということになるのです。
計算で求めたエネルギーや原子間距離は、実験で調べた値とはかなり違っています(特にエネルギーの方は1.5倍ぐらい食い違います)。これはまだ近似が粗いためで、相手の原子核の影響なども考えて元の原子の波動関数を修正するなど、いろいろと近似を高める工夫がされています。
原子価結合法のジレンマ
原子価結合法では、「電子をやり取りして隣どうしで結び付く」、という直感的にわかりやすい形で化学結合を説明することができるのですが、ちょっと困った問題も抱えています。その一つが、有機物に見られる炭素の結合です。
炭素原子では、図4にも示しているように、2s軌道に2個、2つの2p軌道にそれぞれ1個の電子が入っています(残りの1個の2p軌道は空っぽ)。原子価結合法で結合を作る際には電子を1個ずつ出し合う必要がありますから、初めから2個の電子で埋まっている軌道や空っぽの軌道は結合に関わることができません。そのため、結合に関われる軌道は2つの2p軌道だけになり、炭素は「手」を2本しか持っていないことになります。となると、例えば炭素1個に水素が結合してできる化合物はCH2になり、H-C-Hの結合の角度は90度でなければなりません(図7)。ところが、ご存知のように炭素には4本の「手」があり、実際の化合物はCH4(メタン)で、その形は正四面体です。この矛盾をどう考えたらよいのでしょうか。
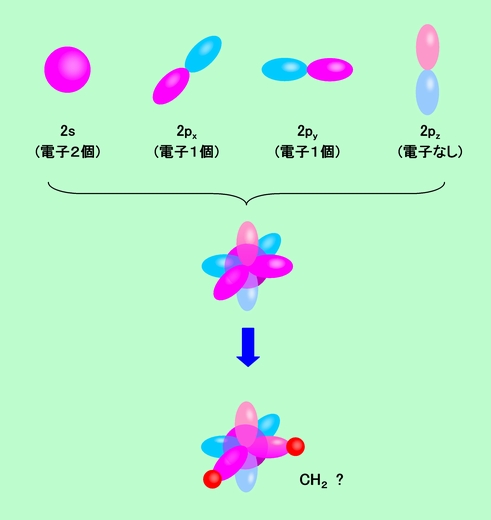
図7 メタンはCH2?
ここで編み出されたのが、「混成軌道」という考え方です。図8のように、s軌道やp軌道をいくつか集めて、新しい軌道に組み替えるのです。前にも書いたように、p軌道も組み換えの結果なのですから、もう一度組み替え直す操作自体には問題はありません。問題があるとすれば、なぜ都合に合わせて変幻自在に組み替えることが許されるのか、ということでしょうが、結果的にエネルギーの低い安定な化合物ができるのであれば、それもアリと考えるのです。
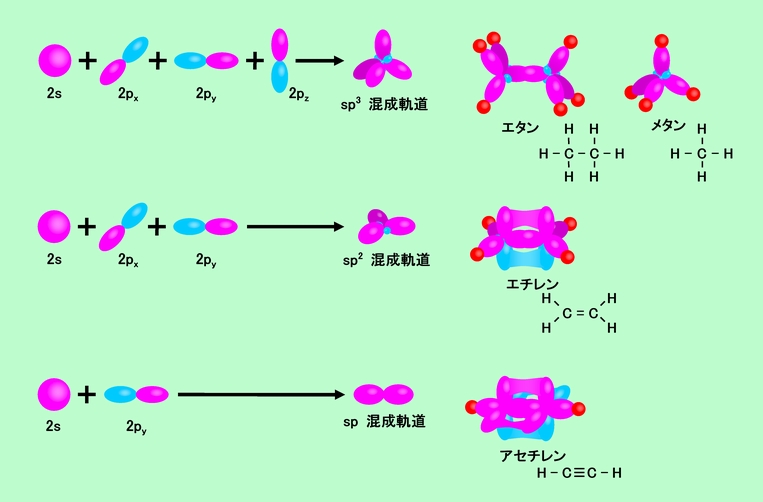
図8 状況に合わせて変幻自在 ― 混成軌道 ―
sp3混成軌道を作るには、s軌道と3個のp軌道全てを使います。その結果、正四面体型に配置された全く同等な4個の軌道ができることになります。4個の電子は各軌道に1個ずつ入りますから、4個のsp3混成軌道の全てが結合に関わることができ、図7のところで問題になった「手」の数やメタンの分子構造も矛盾なく説明できるのです。
s軌道と2個のp軌道を組み合わせると、正三角形に配置した3個の軌道、sp2混成軌道ができます。4個の電子は、新しくできた3個のsp2混成軌道と、余った1個のp軌道に1個ずつ入りますから、やはり4本の「手」を確保することができます。このsp2混成軌道を使って作られるのが、図に示したエチレン分子です。3個のsp2混成軌道のうち2個は水素と結合するのに使われ、他の1個は別の炭素と結合するのに使われます。またp軌道も炭素どうしの結合に使われるので、炭素と炭素は2本の結合でつながることになります(二重結合)。
もう一つ、sp混成軌道というのもあります。今度はs軌道とp軌道1個の組み合わせですから、180度反対向きの2個の軌道になります。この形で作れらる代表的な分子がアセチレンで、余った2個のp軌道も炭素間で結合を作りますから、sp混成軌道と合わせて3本の結合(三重結合)ができることになります。
このように混成軌道の考え方を使うと、炭素の結合の仕方に関する問題を見事に避けることができます。しかし、まだ難題がありました。このままでは、ベンゼンなどの芳香族とよばれる物質の性質をうまく表現することができないのです。
ベンゼンは6個の炭素がつながって輪を作った化合物です。先の混成軌道の考え方を使うと、sp2混成軌道のうちの2個で炭素どうしが環状につながれば(残りの1個は水素と結合)、六角形の構造は説明できます。問題は各炭素に1個ずつ残るp軌道です。普通に考えれば、エチレンと同じように隣の炭素のp軌道とつながって二重結合を作ることになるでしょう。この場合、p軌道の結合の「手」は1本ですから、右隣と結合を作れば左隣とは結合できません。そうすると、六角形の6つの辺のうち、一個おきに3個が二重結合、残り3個が単結合になるはずです。ところが、ベンゼンの性質をいろいろ調べてみると、6個のC-C結合は全て同等で、エチレンの二重結合とは全く違った性質を持っていることがわかります。またまた大きな矛盾が発生するのです。
この問題を解決するために考え出されたのが「共鳴説」です。ベンゼンのp軌道の結合は一箇所に固定されているのではなく、絶えず隣と入れ替わっている、と考えるのです(図9)。
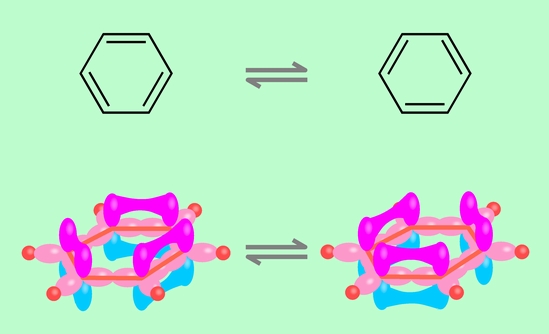
図9 ベンゼンの共鳴構造
このような「共鳴」は、ベンゼンなどの芳香族以外でもいくつか知られています。構造の入れ替わりが十分に速ければ、外から見た時に平均化されて全部の結合が同じに見える、というわけです。
こうした方法によって原子価結合法の問題点を解消して行ったわけですが、混成軌道にしても共鳴にしても、ちょっと苦し紛れ、という感じがしないでもありません。それでも、原子価結合法は直感的に理解しやすいですから、細かな計算はともかく、分子の構造を大まかに見積もる場合には便利です。特に有機分子の構造を考える時は、次に説明する分子軌道法を使うのは大変で、イメージを作りやすい混成軌道の考え方がよく利用されます。
分子全体に広がる軌道で結合する ― 分子軌道法 ―
原子価結合法と並ぶもう一つの方法が分子軌道法です。分子軌道法では、分子の中の電子の波動関数(軌道)として、特定の原子に所属する軌道ではなく、分子全体に大きく広がった軌道を考えます。これが「分子軌道」で、この分子軌道に電子が入って分子全体に広がって飛び回ることで結合ができると考えるのです。もちろん、原子核も電子も複数ある状況で分子軌道を厳密に計算することはできませんから、例によって近似をしてやらなければなりません。
ある原子の近くに電子がいる時には、分子軌道はその原子の原子軌道とよく似た形に見えるでしょう。また別の原子の近くに電子が来た時には、その別の原子の原子軌道に似た形になると考えられます。となると、分子軌道は原子軌道の寄せ集めと見なすことができそうですね。実際に、近似的に分子軌道を求める最も簡単な方法は、既にわかっている個々の原子の原子軌道を足し合わせることなのです。前にp軌道やd軌道で波動関数の組み替えをやりましたが、ちょうどアレと同じように、原子軌道の組み替えで分子軌道を求めるわけで、元の軌道が2個ならば2個、5個ならば5個の分子軌道ができて来ます。その様子を水素分子について示したのが図10です。
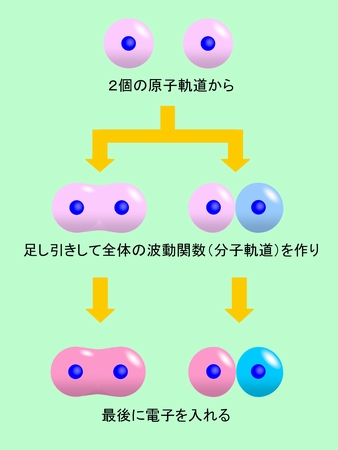
図10 分子軌道法では、分子全体に広がる軌道に電子が入る
スタートは個々の水素原子の原子軌道です。この原子軌道を足し合わせることで、新たに分子全体に広がる分子軌道を作るのですが、その時には、それぞれの原子軌道に入っている電子のことは考える必要はありません。水素原子の原子軌道と同じように、分子全体で1個の電子を考えればいいのです。
この時点では、原子軌道をどういう割合で足し合わせるかはまだ決まっていません。そこで、この未確定要素を含んだ波動関数を、例によってシュレーディンガーの波動方程式に放り込み、エネルギーが最も小さくなるように波動関数を決めて行きます。どこかで聞いたような手順ですね。そうです。原子価結合法の計算方法とほとんど同じなのです。計算手順がよく似ていますから、分子軌道法での計算結果も原子価結合法とよく似た形になり、元の原子軌道を単純に足し算した形と、符合を変えて足し算した(つまり引き算した)形とがセットで出て来ます。ただし、足し合わせる元の波動関数は全く別モノですから、注意が必要です(図5と図10を比べてみてください)。
原子軌道を足し算すると、符号が同じ(図では色が同じ)なので、山と山、谷と谷がうまく重なり、全体で一つの塊のようになります。軌道の形で言えば、2個の餅がペチャっとくっ付いたような状態です。これに対して引き算の方は、符号が逆になって山と谷が重なりますので、2個の原子の中間では両者が打ち消し合って波動関数がゼロ、つまり電子が入れない状態ができます。軌道の形で言うと、2個のゴムボールが押し合っているような感じでしょう。そして、足し算した軌道のエネルギーは元の原子軌道よりも低く、引き算した軌道のエネルギーは高くなります。この状態を、「2つの分子軌道に分裂する」という言い方で表現することもあります。(分子軌道法でも、本当は図6のように原子間の距離によってエネルギーは変化しますが、ここでは距離は固定して考えています)。
最後に、こうして得られた分子軌道に電子を入れて行けば分子が完成します。その時、エネルギーの低い軌道に電子が入れば全体のエネルギーが下がって安定になりますし、エネルギーの高い軌道に電子が入れば不安定になりますから、当然、電子はエネルギーの低い軌道に優先的に入ることになります。ただし、「同じ軌道にはスピンを逆にした2個の電子しか入れない」という規則は、分子軌道にもそのまま当てはまります。
水素分子の場合は図11に示すような電子配置になります。両端の原子軌道が組み合わさって、中央の2つの分子軌道ができ、そのうちエネルギーの低い方の軌道に電子が2個入るのです。
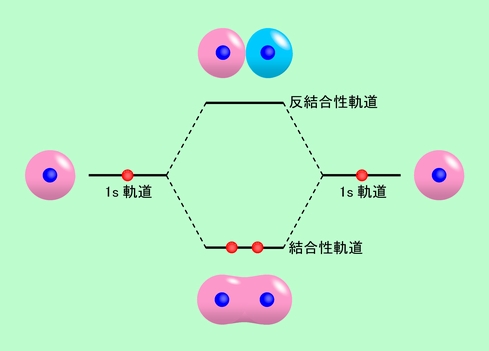
図11 分子軌道法で求めた水素分子の電子の状態
エネルギーの低い分子軌道に電子が2個入ると、元の原子の状態よりも安定になりますから、水素原子はしっかり結合して分子を作ります。しかしエネルギーの高い軌道に電子が入った場合には、結合を作らないで元の原子の状態に戻った方が有利になります。その意味で、エネルギーが低い方の分子軌道は「結合性軌道」、エネルギーが高い方の分子軌道は「反結合性軌道」と呼ばれます。図で見ても、結合性軌道は同じ色の原子軌道が融合してしっかりつながった形、反結合性軌道は違う色の原子軌道が反目し合っているような形になっていますから、直感的に理解できると思います。
分子軌道法のイメージとしてはこんな感じで、分子中の電子の状態を大まかに見積もるだけならばこれで十分でしょう。しかし、分子全体の波動関数をきちんと求めようとすれば、電子をたくさん持った原子の場合と同じように、個々の電子の波動関数を組み合わせた状態を考える必要があります。電子が10個あれば、10個の波動関数を組み合わせるのです。ここで詳しい説明はしませんが、このような複数の電子を考える過程で、原子価結合法と分子軌道法の大きな違いが見えて来るので、そのことにだけ簡単に触れておきましょう。
2個の原子が結合した分子があるとします。原子価結合法では、2個の電子が元の所属のままの状態と、入れ替わった状態とを考えました。この時、2個の電子は必ず2個の原子核に1個ずつ分配されます。どちらかに電子が偏った状態というのは、初めから想定されていません。これに対して分子軌道法では、別々の原子の原子軌道を組み合わせて分子軌道を作ります。そこに電子を入れるということは、元になったそれぞれの原子軌道に平等に電子が分配される、ということを意味します。この場合、電子が元通りの所属に収まった状態と、電子が入れ替わった状態だけでなく、一方の原子に電子が2個とも集まった状態も同じように起こり得ることになります。つまり分子軌道法では、原子価結合法では全く想定されていなかった、電子が偏った状態(イオンの状態)も含めて考えていることになるのです。言い換えると、原子価結合法では共有結合のみを考え、分子軌道法では、共有結合とイオン結合が半々の状態を考えている、ということです。
それでは実際の化学結合はどうかというと、普通に共有結合として扱われている結合の大半はこの中間でしょう。水素や窒素のように同じ原子がつながった分子の場合はほとんどイオン性はありませんが、原子の種類が違う場合や、同じ原子でも(それに結合している原子や原子団が違うなど)周りの環境が違っている場合は、いくらかイオン性が出て来ると考えられます。このような現実の分子と比べると、原子価結合法と分子軌道法は、その両極端の近似法であると言えます。ただし現在では改良がいろいろと進んで、様々な条件を取り込めるようになっていますから、極端な近似ではなく、もっと現実的な答が出せるようになっています。
いろいろな分子軌道
原子軌道にはs軌道だけではなくp軌道やd軌道などもありますから、これらを組み合わせて作られる分子軌道にも実に様々なものがあります。また分子軌道作りに参加する原子軌道の数も、2つだけとは限りません。複雑な分子になれば、何個もの原子の軌道が集まって、たくさんの分子軌道を作り出します。これらをいちいち紹介していたらキリがありませんので、ここではs軌道またはp軌道が2つ関わる場合に絞って、どのような分子軌道ができるのかを見てみましょう。もちろんこれらの分子軌道はシュレーディンガーの波動方程式を解くことで出て来るのですが、図12のように原子軌道の形を見比べるだけでも、おおよそ見当をつけることができます。
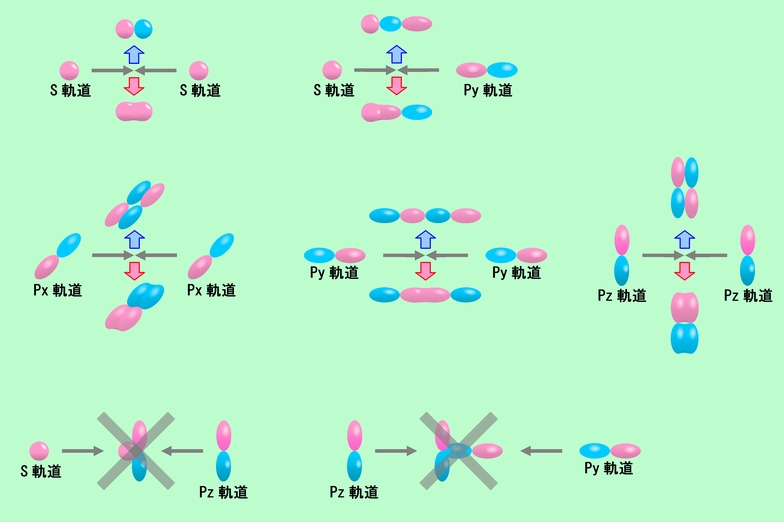
図12 原子軌道の組み合わせいろいろ
図の左上のs軌道どうしの結合は、水素の場合と同じですから、改めて説明する必要はないでしょう。次にs軌道とpy軌道の組み合わせを見てみると、s軌道の球とpy軌道のダンベルの片方とが交わる形で、符号が同じもの(赤と赤、または青と青)が重なった結合性軌道と、符号が逆のもの(赤と青)が接した反結合性軌道を作ることができます(図の右上)。これらは原子軌道が頭と頭を突き合わせていて、結合の方向(図では左右方向)から見た時にs軌道と同じような丸い形に見えますから、「s」に対応するギリシャ文字の「σ(シグマ)」を使って「σ結合」、できた分子軌道を「σ軌道」と呼んだりします。
それではp軌道の場合はどうでしょうか。この場合も、同じ方向のものどうし、つまりpxとpx、pyとpy、pzとpzならば、符号の同じもの(赤と赤、または青と青)が組み合わされば結合性軌道、符号が逆のもの(赤と青)が組み合わされば反結合性軌道ができます(図の中段)。この中でpy軌道どうしの組み合わせは頭と頭の突合せで、横から見ると丸く見えますのでσ結合です。これに対してpx軌道どうし、pz軌道どうしの組み合わせでは横に並ぶ形ですから、横から見てもp軌道そのもののダンベル型ですね。そのためこのような結合は、「p」に対応するギリシャ文字の「π(パイ)」を使って「π結合」、できた分子軌道は「π軌道」と呼ばれます。
ところが、例えば図の右下のようにpy軌道とpz軌道を組み合わせようとすると、重なっている部分の半分が同符号(赤+赤)、残りの半分が逆符号(赤+青)という形になってしまいます。これでは結合性軌道にも反結合性軌道にもなれませんから、この組み合わせはあり得ません。px軌道とpy軌道、px軌道とpz軌道の組み合わせも同様で、お互いに全く噛み合わない間柄なのです。同じようなことは、違うタイプの軌道間でも起こります。s軌道とpx軌道や、s軌道とpz軌道の場合がそうで、図の左下に示すように、やはり結合性軌道にも反結合性軌道にもなれません。
このように、いろいろな原子軌道が参加して分子軌道を作ると言っても、中には全く参加できない組み合わせもあるわけです。また、分子軌道作りに参加する場合でも、その関わり合いの深さは原子軌道によって様々です。中心メンバーとして大きく関わる場合もあれば、ちょっと加わる程度、という場合もあるのです。さらに、関わり合いが深ければ深いほど、結合性軌道と反結合性軌道のエネルギーの分裂が大きくなる、という特徴もあります。一般にはσ結合の方がπ結合よりも関わりが深く、エネルギーが大きく分裂します。
これらの関わり合いの深さを決めるのは、原子軌道がしっかり重なり合うような形をしているかどうかと、エネルギーが近いかどうかです。図12にも示しているように、原子軌道がしっかり重ならなければ、一緒になって分子軌道を作ることはできません。また、形の上ではちゃんと重なるように見えても、例えば1s軌道と2s軌道ではエネルギーがかなり違いますから、うまく共同作業することができないのです。
それでは、いくつかの分子の分子軌道を具体的に見てみましょう。まず、水素の次に簡単なヘリウムです。水素分子の場合は、図11のように結合性軌道に2個の電子がキッチリ収まって安定状態になっています。これに対して、同じように1s軌道の足し合わせで分子軌道を作ることができるヘリウムの場合は、電子が4個あるために、水素のようには行きません。結合性軌道は2個の電子で満員ですから、残りの2個は反結合性軌道に入るしかないからです。こうなるとエネルギーは差し引きゼロで、分子を作っても何の得もありませんので、ヘリウムでは水素のような安定な分子はできない、ということがわかります。
次に、代表的な2原子分子である窒素と酸素です。窒素と酸素の分子軌道は図13のようになります。(ただし、全体が見やすいように、エネルギーを示す線の位置を少し動かしていますので、エネルギーの値そのものは正確ではありません)。
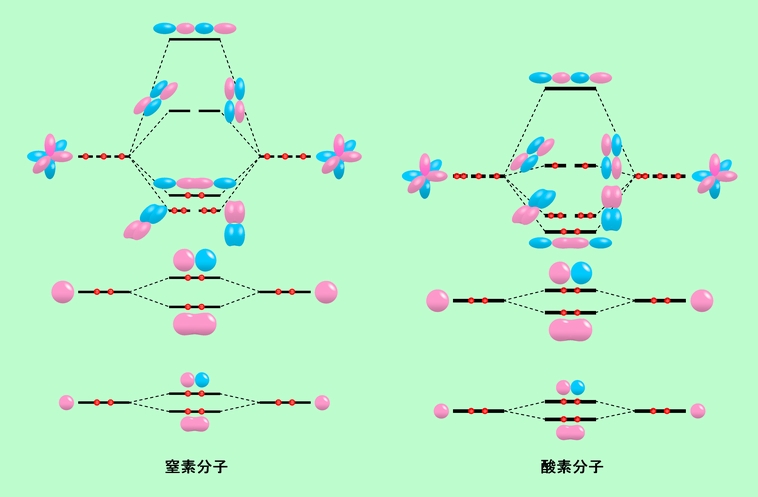
図13 窒素と酸素の分子軌道
図11の水素の例と同じように、原子軌道が合わさって分子軌道に分裂する様子が破線で描かれていますが、これはあくまでも主に関わる原子軌道を示しているに過ぎません。それぞれの分子軌道には破線で結んだ以外の原子軌道も一部参加していますし、さらに分子軌道どうしの相互作用もあって、実際にはけっこう複雑です。このような複雑な相互作用の一つが、下から5番目と6番目の分子軌道(σ軌道とπ軌道)の位置関係の逆転に現われています。この逆転が起こる原因は、2py軌道からできるσ軌道と、その下の2s軌道からできるσ軌道の相互作用にあります。窒素の場合はこの相互作用が強く、2py軌道からできるσ軌道のエネルギーがπ軌道よりも高くなる(もちろん、反対に2s軌道からできるσ軌道のエネルギーは低くなる)のに対して、相互作用の弱い酸素の場合は、σ軌道のエネルギーがπ軌道よりも低いままになるのです。
窒素分子と酸素分子のもう一つの大きな違いは、電子の配置です。窒素では2p軌道が関わる分子軌道のうち、3個の結合性軌道がちょうど全部埋まる形になっています。原子価的な言い方をすれば結合が3本、つまり三重結合になっているわけです(1s軌道と2s軌道からできる分子軌道にも電子がありますが、結合性軌道と反結合性軌道の両方に入っているために帳消しになってしまいます)。これに対して酸素分子では、窒素分子よりも電子が2個多いですから、これが上から2番目の反結合性π軌道に1個ずつ入ることになります。これによって3個の結合のうちの1個分が帳消しになって、酸素分子の結合は2本になります。また、反結合性π軌道に入っている2個の電子は、同じ軌道でスピンの向きを逆にしてペアを作っている他の電子と違って、スピンの向きが同じで1個ずつ孤立しています。そのため酸素分子は窒素分子と比べて反応性が高いですし、磁石に引き寄せられる、という特徴的な性質も持っているのです。
続いて、原子価結合法では説明に苦労した有機分子を見てみましょう。分子軌道法を使うと、原子価結合法で無理やりひねり出した混成軌道や共鳴の考え方は必要ありません。例えばメタンの分子の場合、炭素の4個の軌道(2s、2px、2py、2pz)と4個の水素の1s軌道、計8個の原子軌道から、8個の分子軌道が作られます。このうち4個は結合性軌道、4個は反結合性軌道ですが、結合性軌道の概観はおよそ図14のようになります。

図14 メタンの分子軌道(結合性軌道)
左端は分子全体に広がる軌道、他の3つは水素を2個ずつ組にしたような形の軌道で、雰囲気としては、左端の軌道がs軌道、他の3つがp軌道に似た形と言えるでしょう。4つの分子軌道を合わせて考えると、4個の水素原子の周りの環境はどれも全く同じであることがわかると思います。つまりCとHの関係は全て同等、というメタンの性質がちゃんと表現されているのです。ついでに言うと、メタンの電子にはエネルギー的に2種類あり、その数の比率が1:3であることがわかっています。これは図14の左端の軌道と右の3つの軌道に対応しており、分子軌道法でうまく説明できるのです。
ベンゼンの構造についても、分子軌道法ならば何の問題も発生しません。炭素の6個の1s軌道と6個の2s軌道、18個の2p軌道、それに6個の水素の1s軌道が組み合わさって、分子全体に広がる36個の分子軌道ができるわけで、部分的に二重結合だとか単結合だとかを考える必要はないのです。図15に、そのいくつかの分子軌道を示しておきました。
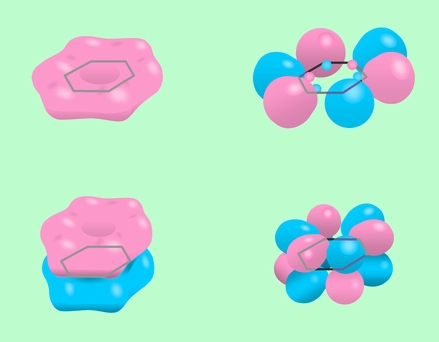
図15 ベンゼンの分子軌道の例
原子価結合法と分子軌道法の合わせ技
原子価結合法では、実際の分子をうまく説明するために混成軌道やら共鳴やらの複雑な考え方を取り入れる必要があり、ちょっと無理をしているように見えます。それでも、隣の原子との関係だけを考えればいいわけですから直感的に理解しやすく、特に有機化学の分野で盛んに使われています。この場合、エネルギーなどを細かく計算するというよりも、分子の構造をイメージする、という使い方が主でしょう。一方、分子軌道法は、直感的に分子構造をイメージするのは苦手です。全貌を捕まえようとすれば多くの原子の軌道を寄せ集めなければならないのですが、原子の数が増えると計算量が膨大になってしまうのです。しかし理論の組み立て自体はより自然な形になっていると言えますから、コンピューターが発達した現代では、計算化学にはもっぱら分子軌道法が使われているようです。とは言うものの、誰もが自由にスーパーコンピューターを使えるわけではありませんし、精度のよい計算をしようとすれば、それなりに時間も労力もお金もかかります。そこで、細かな数値はともかく、大まかに分子の性質を計算して調べようとする場合には、両方の考え方をうまく組み合わせた方法もよく使われます。
例えば、何度も登場するベンゼンですが、本格的に分子軌道法で計算するには、全部で36個の軌道を扱わなければなりません(専門にやっている人から見れば、この程度は大したことではないのでしょうが)。そこで混成軌道の考え方を拝借して、炭素のsp3混成軌道と水素の1s軌道を使って分子の基本的な骨格部分(正六角形の部分)を作ってしまい、残った6個のpz軌道だけを分子軌道法で扱う、という荒技が考えられました。この場合pz軌道は横に並んでいるわけですから、できる分子軌道は全てπ軌道になります。さらに、このようにπ軌道が分子全体に広がっているような分子では、計算の過程で、もっと大胆な近似をする場合もあります。関係する軌道全部の相互作用を生真面目に計算するのではなく、隣り合った炭素間の重なり以外は無視してしまう、といった方法です。このようにして、とことん簡略化して求めたベンゼンの分子軌道を示したのが図16です。元々6個のpz軌道から作ったのですから、分子軌道はここに示した6個で全部で、下から三つ目の軌道までが電子で埋められています。
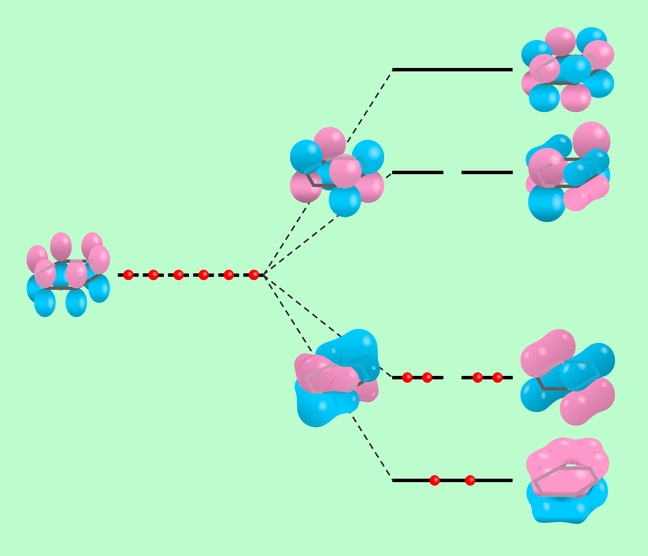
図16 pz軌道だけで作ったベンゼンの分子軌道
ずいぶん大胆な近似をした割には的を射た答が出て来るのが面白いところで、図16の6個の分子軌道は、キチンと計算した時の36個の中にもちゃんと現われます。36個のうち主にpz軌道から組み立てられる6個の軌道とほとんど同じ形になり(図15の下段の2つはその例です)、しかも電子で埋まった軌道と空っぽの軌道の関係まで一致するのです。
分子の性質、特に化学反応に関わる性質に大きく影響するのは、他の分子との間で電子のやり取りをしやすい分子軌道です。これに相当するのが、電子の入った軌道の中で最もエネルギーが高い分子軌道と、空の軌道の中で最もエネルギーが低い分子軌道で、図16ではこのような重要な軌道の状況がほぼ再現できているわけです。
雑科学ホーム
hr-inoueホーム