
雑科学ホーム
hr-inoueホーム
● 表面分析の話 ●
表面の意味
宇宙空間のようなよくわからないものは別にして、物には必ず「端」があり、「表面」があります。そしていろいろな意味で、「表面」は内部とは違っています。
まず、物の形というのは表面の形のことです。中が空洞であろうがアンコが詰まっていようが、形そのものには関係ありません。形を決めるのは表面です。次に、内部にある原子や分子は同じ仲間に囲まれていますが、表面の原子や分子は半分が別の物に接していますから、エネルギーが高い、不安定な状態になっています。もし、他の原子や分子と接している方が安定ならば、半分と言わず、周囲全部を他の分子で取り囲んでもらった方が得なので、元の物体に留まっていないで、別の側に溶け出してしまうはずです(こうなると2つの物は分離していられなくなりますから、表面はなくなってしまいます)。また、表面は様々な変化を起こす基点になります。その物体を代表して外部と接しているのですから、外からの攻撃を受けるのも、外へ働きかけるのも、全て表面の担当です。このように、外から見た時のその物体のとりあえずの性質を決めるのは表面なのです。
表面の性質は、物が小さくなればなるほど、薄くなればなるほど、その重要度を増します。言うまでもないことですが、小さくなるほど表面の割合が大きくなるからです。図1に、1cm角の立方体を分割して行った時の表面積の変化を示しました。青線はタテ、ヨコ、高さ方向に等分に分割した場合、赤線は薄くスライスして行った場合です。
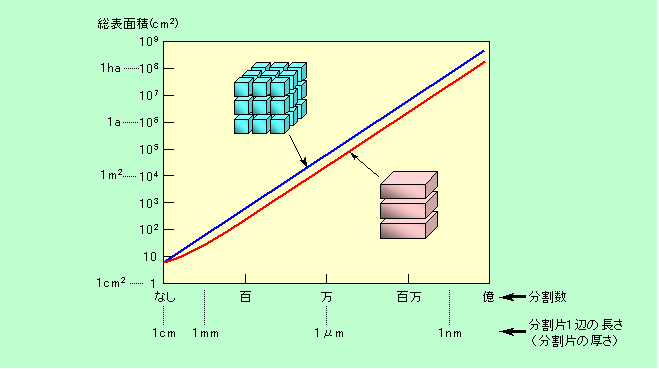
図1 立方体を分割した時の表面積の増加
図では縦軸、横軸共に対数目盛りで表示していますが、普通に目盛った場合には、どちらも完全な直線になります。分割を進めると急激に面積が増えることを期待していたかもしれませんが、実際には一辺の長さが半分になっても面積は2倍にしかなりません。とは言え、角砂糖程度の大きさのものが、1万分割して一辺1μmになるとタタミ四畳分の6m2(スライス型でも2m2)になり、そのさらに1000分の1の1nmまで分割すると1ha(ヘクタール)に迫るレベルになるわけですから、なかなか大したものです。また、1辺1nmとなると数十個分の原子の大きさしかありませんから、全体の七割ほどの原子は表面に出ていることになります。ほとんど表面ばかり、という状態なのです。
よく粉状の物質の特徴を表すのに1g当たりの表面積(比表面積)というのが出て来ますが、例えば密度が3g/cm3ぐらい(普通の石ころはこんなものです)の物質の場合、大ざっぱに言って大きさ1μmの粒子であれば1g当たりの表面積は2m2程度と覚えておけばよいでしょう。ちなみに、普通の人が粉末を指先で触ってザラザラした感触がする場合の粒子の大きさは最小10μmぐらいと言われていますから、ザラザラ感がないスベスベした感触の粉は10μmよりも小さく、m2級の比表面積を持っていることになります。
表面を「見る」方法
このように薄膜や粉末で特に重要で、物の顔とも言える表面ですが、その「顔」の細かな「表情」を見るにはどのようにすればよいでしょうか? ここで言う「表情」というのは、もちろん色や形だけを指すものではありません。もっとミクロな原子・分子の種類や配列、化学的な状態、外部からの他の物質の吸着なども含んだもので、それらをどうやって分析するか、ということです。
表面分析に限らず、何かを分析しようとすれば、試料から出て来る何らかの信号を捕まえることが必要です。信号を出すにはエネルギーが必要ですが、残念ながら生物以外では自発的に信号を出すようなものはほとんどありません。自分で壊れて放射線を出す放射性元素などが唯一の例外でしょうか。そこで、試料から信号を出させるには、外から刺激(エネルギー)を与えてやる必要があります。つまり、何かでつついて応答を見る、ということです。この「何でつつくか」、ということと、そこから出て来る「どんな応答を見るか」、ということで、いろいろな分析方法が出てくるわけです。このような観点から、代表的な表面分析の方法をまとめたのが下の表です。もちろん分析手法の種類はこんなものではありませんが、とりあえず、こういう見方もできるという例として見てください。
表 刺激と応答の種類で見た表面分析法いろいろ
| 刺激\応答 |
赤外線 |
可視光線 |
紫外線 |
X線 |
電子線 |
イオン線 |
| 赤外線 |
赤外線吸収分光 |
|
|
|
|
|
| 可視光線 |
|
光学顕微鏡
蛍光分光
ラマン分光
エリプソメトリー |
|
|
|
|
| 紫外線 |
|
蛍光分光 |
紫外線吸収スペクトル |
|
紫外線光電子分光 |
|
| X線 |
|
|
|
X線回折
蛍光X線分光 |
X線光電子分光 |
|
| 電子 |
|
|
|
電子線プローブX線分光 |
電子顕微鏡
オージェ電子分光 |
|
| イオン線 |
|
|
|
|
二次イオン顕微鏡 |
二次イオン質量分析 |
刺激として与えるものがどの程度の深さまで侵入するか、ということと、応答として出て来る信号がどの程度の深さから脱出可能か、ということで、その方法で分析できる表面からの深さが決まります。一般的に言って、透過力の強いX線はかなり奥まで侵入しますし、かなり奥からも出て来ることができますが、電子線やイオン線はごく浅いところの情報しか拾いません。また、紫外線や可視光線は(相手の物質にもよりますが)表面附近のみ、一方、赤外線はやや深いところの情報も運んで来ることが多い、と考えてよいでしょう。
表に挙げたものの他に、ガンマ線を用いるメスバウアー分光や、中性子線を使った回折法、赤外線の吸収を熱膨張に伴う振動(つまり「音」)として捕らえる光音響分光などもあります。また、走査トンネル顕微鏡や原子間力顕微鏡などのプローブ顕微鏡も、前表のような分類の仕方をするのは難しいですが、有力な表面分析手段のひとつです。これらの分析手法について一つ一つ解説すると膨大な量になってしまいますから、ここでは表に挙げたものに絞って、ごく簡単に見てみることにします。
電磁波の分光
赤外線や可視光線、紫外線の分光というのは、光を当ててどの波長が吸収されるかを調べるもので、表面からの反射光を対象にすれば表面分析の手法となります。プリズムや回折格子を使って光を波長(つまりエネルギー)成分に分けて強度を調べますので「分光」と呼ばれるのです。
赤外線は分子の中の化学結合を振動させる性質があるので、吸収される波長によって化学結合の種類が調べられますし、紫外線は分子の中の電子に吸収されますから、電子の状態がわかります。これらの情報から、主に有機分子の化学構造が推定できるのです。可視光線も紫外線と同じようなものですが、紫外線よりもエネルギーが小さいので、ごく限られた状態の電子にしか吸収されません。ただし、可視光線を吸収するということは人間の目に色が見えるということですから、こういう分子があるとよく目立ちます。光学顕微鏡も、この色の変化をかなりの部分で頼りにしています(
顕微鏡の話参照)。蛍光分光は、紫外線や可視光線を一旦吸収した後に、より波長の長い別の光を出す様子を調べるもので、やはり分子構造に関する情報が得られます。無機物はあまり蛍光を出さないことから、未知の物が有機物なのか無機物なのかを調べる場合にも有効で、
発光の話でも書いているように、昔の絵画に使われている絵の具を調べるのにもよく利用されています。ラマン分光はちょっと毛色が違っていて、レーザーなどの強力な光を当てると、その光のエネルギーの一部が試料分子の振動に使われて吸収されたり、逆に分子の振動からエネルギーをもらったりして、波長が少しずれた弱い光を散乱する現象(ラマン散乱)を利用した分析方法です。分子の振動に必要なエネルギーが差引かれたり付け足されたりしますから、元の光の波長からのずれは、ちょうどその結合が吸収する赤外線のエネルギーに相当しています。つまり、利用する光は可視光線でも、取り出される情報は赤外線の吸収と同じもの、ということです(実際には化学結合の種類によって赤外線吸収は強くてもラマン散乱は弱い場合やその逆の場合があるので、両者は使い分けられています)。
ここで、赤外分光などでよく目にする「フーリエ変換」についてちょっと触れておきましょう。
Fourier
Transformation = FTと略されるヤツです。昔の分光法と言えば、先にも書いたようにプリズムや回折格子を使って光を波長に分け、それを端から順に、あるいは検出器をズラッと並べていっぺんに強度測定していました。プリズムなどを回転させて順に調べて行く方法では測定に時間がかかりますし、検出器を並べる方法では、解像度に限界があります。そこで、全部の波長を含んだ光を同時に当てて、フーリエ変換の原理を使って波長ごとに分析する効率のよい方法が登場して来たのです。では、いろいろな波長の光が混ざった状態から、どうやって特定の波長の光の強度を知ることができるのでしょうか。それをこれから簡単に説明しましょう。
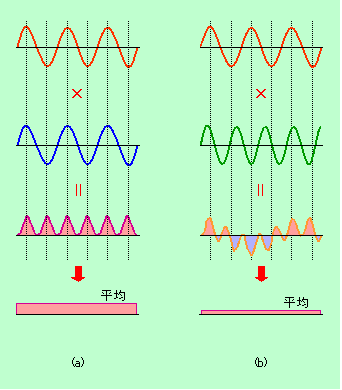
図2 波どうしの掛け合わせ
図2(a)のように、ある周期の波に、それと同じ周期の別の波を掛け合わせてみましょう(足し算ではなく、掛け算にします)。すると、山と山が単純に掛け合わされて大きな山になると同時に、谷と谷もマイナスどうしが掛け合わされてプラスの大きな山になりますから、全体を平均すると、プラスの大きな値になります。これに対して(b)のように周期が違う波を掛け合わせる場合はどうでしょうか。今度はどう動かしても山や谷の位置がずれてしまいますので、掛け合せの結果はプラスになったりマイナスになったりして、平均すると小さな値になってしまいます。この理屈を応用すれば、図3のように、いろいろな周期が入り混じった複雑な形の波から、特定の周期成分を取り出すことができるのです。
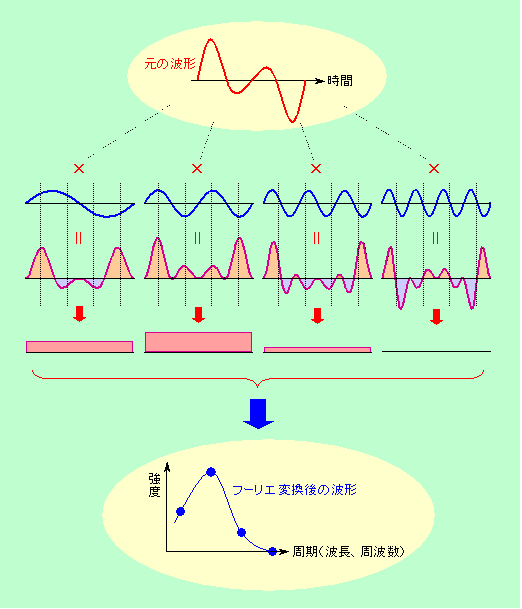
図3 特定の周波数成分だけを強調して取り出すフーリエ変換
赤色で示したように時間変化する元の波に、青色で示したいろいろな周期の波を掛け合わせます。掛け合わせる波と同じ周期の成分が元の波にも含まれていれば、プラスどうし、マイナスどうしの掛け算になる可能性が高くなりますので、図2(a)と同じように掛け合わせの結果はプラスになる部分が多くなります。逆に、掛け合わせる波の周期が元の波に含まれていなければ、図2(b)と同じように掛け合わせの結果はプラスになったりマイナスになったりすることになります。掛け合わせた結果を平らにならして平均化すれば一目瞭然。元の波にその周期の成分が含まれていれば平均値は大きく、あまり含まれていなければ平均値は小さくなるのです。図の例の場合ですと、元の波には左から2番目の周期成分が最も多く含まれ、左端の周期成分はその半分、そして右端の周期成分はほとんど含まれていない、ということになりますから、周期を横軸に、強度を縦軸にとってプロットし直すと、一番下に示したようなパターンが描けることになります。このような操作を数学的にやるのがフーリエ変換です。
それでは、実際の分光測定ではフーリエ変換をどのようにして取り入れているのでしょうか。光は元々波の性質を持っていますから、これをそのまま利用したいところですが、実はそうは行きません。光の波そのものは電場や磁場の振動なのですが、山の部分が今通過したとか、ここに谷の部分が来たとかは、直接観察することができないのです。観測可能なのは光の強度(波の振れ幅=振幅)のみ。というわけで、フーリエ変換を使おうとすれば、電場や磁場の波の情報を、観測可能な強度の情報に変換してやることが必要になります。それには、光の干渉を利用した図4のような仕組みが使われます。
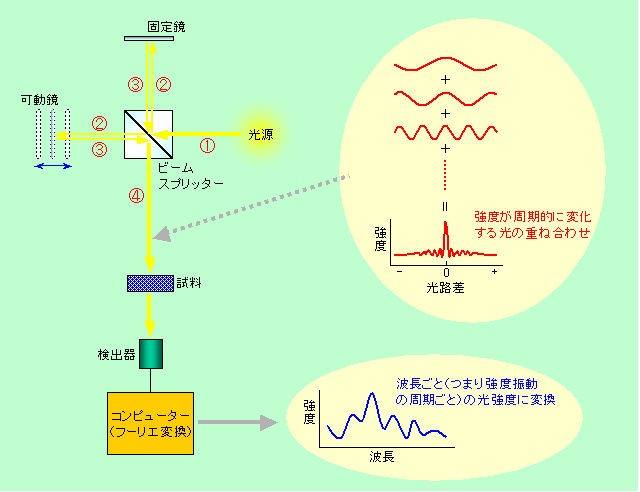
図4 フーリエ変換を使えるように工夫された分光装置
いろいろな波長が混じった光は、半分透過して半分反射する鏡(ビームスプリッター)によって2つに分けられます。その一方は普通の鏡で反射されて戻って来ますが、もう一方は前後に動く鏡(可動鏡)で反射されて戻って来ます。これら2つの光がビームスプリッターのところで再び一緒になるのですが、一方の光は可動鏡の位置によって通って来た距離(光路長)が変化していますから、両者の光路長に差ができることになります。光路差が全くない場合は、常に山と山、谷と谷が重なりますから、2つの光は強め合います。これに対して波長の半分の光路差ができると、山と谷が重なるので、光の強度はゼロになり、さらにずれが大きくなって光路差が一波長分になると、2つの光は再びピッタリ重なってまた強くなります。このように、可動鏡の移動に伴って、2つの光が干渉して強め合ったり弱め合ったりを繰り返し、強度が振動する光が作られるのです。すぐにわかると思いますが、光が強くなったり弱くなったりする周期は、元の光の波長によって違います。例えば波長が0.1mmの光は、光路差が1cm変わる(可動鏡が5mm動く)間に100回振動しますが、波長0.2mmの光は50回しか振動しない、という具合です。つまり、元の光に含まれている様々な波長の成分は、それぞれが違った周期で強度振動するようになるのです。ただし、いろいろな光が重ね合わされていますから、このままでは図4に赤で示した波のようにグチャグチャで何が何だかわかりません。パッと見て目立つ特徴と言えば、波長に関係なく強め合う光路差ゼロのところで強度が最も強くなることぐらいで、それ以外の情報は、人がどんなに一生懸命に眺めても出てこないでしょう。そこでフーリエ変換が登場します。図3で説明したように、このグチャグチャな信号にいろいろな周期の波を掛け合わせてやれば、それぞれの周期成分がどのくらいの割合で含まれているかがわかるのです。どんな周期で振動するかは、先に書いたように元の光の波長で決まっていますから、結局、各波長がどれくらいの強度で含まれていたかがわかる、つまり、プリズムや回折格子と同じように分光ができることになるわけです。
実際の分光装置では、図4のように、いろいろな振動成分が重なった(一見、グチャグチャな)光を試料に当て、試料を通って来た光の強度を検出器で捕まえます。検出器で捕まえられる光も、当然、可動鏡の動きに応じて強度が変化していますから、このデータをフーリエ変換して波長ごとの強度に直し、試料がない時と比べれば、どの波長の光が試料に吸収されたかがわかるのです。このような処理をするには相当な量の計算をこなさなければなりませんから、フーリエ変換を使った分光測定が実用になったのは、コンピューターの発達のおかげ、と言うことができます。
エリプソメトリー
少々聞きなれない名前かもしれませんが、コーティング膜などの分析によく利用されている方法です。普通の光は360度あらゆる方向に振動していますが、エリプソメトリーでは振動の方向に制限がついた「偏光」、特に振動の方向が楕円を描いて変化する「楕円偏光」が使われます(エリプソ(ellipse)とは楕円のことです)。楕円偏光は、互いに垂直に振動する2つの成分が、タイミングを少しずらせて振動することで作られます(タイミングがピッタリ合うと斜め45度の直線偏光に、逆に一方が最大に振れた時に他方がゼロになるタイミングだと円偏光になります)。これを試料に当てて反射させると、膜の表面で反射するだけでなく、膜の表と裏で反射した光が干渉を起こして強め合ったり弱めあったりしますし、膜を通過する光は進行速度が膜の性質(屈折率)に応じて遅くなりますから位相(波の山や谷の位置)もずれます。これらの影響が、膜に平行な振動成分と垂直な振動成分では違うので、2つの成分を合成してできる楕円偏光の状態も、初めに入射した光とは違ったものになります。それを解析することで、膜の厚さや屈折率を知ることができる、というわけです。実際のエリプソメトリーは、原理も光学系もかなり複雑ですから、ここではおよその雰囲気を示す絵だけを示しておきます(図5)。
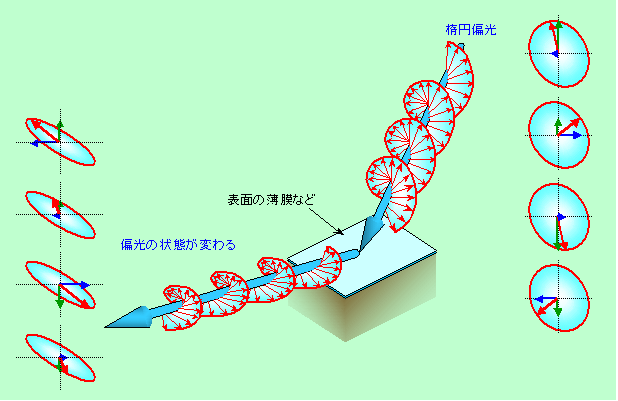
図5 楕円偏光の状態変化を見るエリプソメトリー
X線を検出する分析法
これまでは、いわゆる「普通の光」を使った分析方法でしたが、もっと波長の短いX線を捕まえる分析方法をまとめて見てみましょう。X線の応答を得るための刺激としては、同じX線の場合と電子線の場合とがあります。
X線回折では、入れたX線がそのまま出て来ますから、検出側としては強度を測るだけです。どの方向に強いX線が出てくるかで結晶構造を調べるわけで、その原理は
結晶の話に書いていますし、基本的に表面分析の方法ではありませんので、ここでは説明は省略します。
蛍光X線分光は、刺激は同じX線ですが、試料にX線を当てた時に出て来る、元とは違った波長のX線を分析するものです。ちょうど可視光線や紫外線を当てた時に出る蛍光とよく似ていますから、このX線を「蛍光X線」と呼ぶのです。その発生の原理を大まかに示したのが図6です。

図6 蛍光X線が出る仕組み
初めに当てたX線によって、試料に含まれる原子が持つ電子が叩き出されます。この時、エネルギーが低い電子が叩き出されると、その後にできた空席にエネルギーの高い電子が落ち込み、余ったエネルギーが蛍光X線として放出されます。電子のエネルギー差が小さいと、X線ではなく紫外線や可視光線になりますが、一番下の電子が関与する場合にはエネルギー差が大きいので、高エネルギー(短波長)のX線が出ることになるのです。
原理からわかるように、蛍光X線のエネルギーは元のX線には関係なく、原子が持っている電子のエネルギー状態だけで決まります。この電子のエネルギー状態というのは原子の種類で決まっていますから、蛍光X線のエネルギー(波長)を調べれば、どんな原子が試料に含まれているかを知ることができるのです。特に、重い金属原子からは蛍光X線が強く出ますから、金属の酸化物が主成分である無機物の分析に威力を発揮します。可視光線の蛍光分析が有機物の判定に有効であったこととは対照的ですね。透過力の強いX線を使いますから必ずしも表面の分析とは言えないのですが、X線と言えどもいくらでも深く入り込むわけではなく、表面から数μm程度の情報を見ることになる場合もあります。また、わざと表面ギリギリの浅い角度でX線を当てることで表面数nmの分析をする方法もあります。
電磁波の一種であるX線は、電子線やイオン線と違って途中に空気があってもあまり影響されませんから、試料を普通に空気中に置いた状態で測定できます(X線の中でも波長が1nmを超える低エネルギーのものは空気中での透過力が弱いので、この辺りも含めて高精度に分析できる装置では真空にして測るタイプのものもあります)。装置はそこそこ大掛かりではありますが、試料に特別な細工は必要ありませんので、分析法としては比較的手軽な部類に入ると言えるでしょう。そのため、真空にしたりするとダメージを受けるような水気の多い物質ですとか、貴重な美術品や遺跡からの出土品などの、あまり手を加えられない対象の調査によく使われます。微量成分の分析で産地が特定できたりするのです。また、少し前に、微量に含まれる不純物を調べることで毒物であるヒ素の出所を特定した話がありましたが、これに使われたのも、(ちょっと特殊な)強烈なX線源を用いた蛍光X線分析です。
X線ではなく電子線を試料に当てて、出て来るX線を分析する方法もあります。電子線プローブX線分光と呼ばれる方法で、EPMA(
Electron
Probe
Micro
Analysis)と略されます。X線が出て来る原理は図6の蛍光X線と同じで、電子を叩き出す役目を電子線がする点が違うだけです(ちなみに、先のX線回折装置や蛍光X線分析装置で初めに照射するX線を発生させるのに使われるX線管も、ほとんどが電子線を金属にぶつけてX線を発生させる方式です)。ただ、電子線は空気中では空気の分子に邪魔されて真っ直ぐに飛べませんので、試料を入れる部屋も含めて、装置の内部は真空にしなければなりません。また、電子線はX線ほど透過能力がありませんから、分析できるのは試料のごく表面(深さ1μmぐらい)のみで、典型的な表面分析法の一つです。
EPMAの大きな特徴の一つは狭い範囲の分析ができることです。X線の場合は適当なレンズがないことから、初めに照射する範囲を小さく絞ることができず、普通の蛍光X線の装置では数mmから数cmという広い範囲の分析になってしまいます。これに対して電子線の場合は、電子レンズを使ってμmレベルに絞ることもできますから、まさに顕微鏡で見るように分析することができるのです。実際にEPMAの電子線照射部分の構造は
走査型電子顕微鏡(SEM)そのもので、電子検出器の代わりにX線検出器が付いている、と考えればよいのです。ですから、SEMと同じように電子線を試料上で走査して、出て来る特定の波長のX線を捕まえて行けば、ある成分が試料上でどのように分布しているかを絵にすることもできます。X線を使って顕微鏡画像を作れるわけです(EPMAをそのまま日本語にすれば「電子線を探針にした微細部分析」ですから)。
蛍光X線にしてもEPMAにしても、X線のエネルギー(波長)を分析すること、つまりX線の分光が必要です。しかし、可視光線のようにプリズムで屈折させることはできませんし、普通の回折格子は波長が短かすぎて使えません。それではどうやって分光するのでしょうか。その方法は2つあります。
一つは、X線のエネルギーを電流に変えて直接測定する方法です。半導体でできた検出器にX線が飛び込むと、動きが取れない状態にあった電子がエネルギーをもらって動ける状態に飛び上がり、電流が流れるようになります(
半導体の話参照)。飛び込むX線の波長が短くエネルギーが大きければ、より多くの電子が飛び上がって多くの電流が流れるのです。ここで疑問に思う人がいるかもしれません。「波長が短ければエネルギーが大きいというのはわかるが、波長が長くても強度が大きければ、やはりエネルギーは大きくなるのではないか」と。でも心配はいりません。X線も含めて電磁波というのは、エネルギーの粒のような性質を持っていて、1個1個の粒が、波長に応じたエネルギーを持って飛んで来ます。電磁波は連続して飛んで来るように見えますが、実は1個ずつバラバラになっているのです。そして強度が大きいというのは、実は粒の数が多いということであって、1個1個の粒のエネルギーの大きさとは関係ないのです。そのため、検出器に1個のX線が飛び込むたびに、そのエネルギーに応じた電流がピッと流れ(パルスと言います)、また次のX線が来ると、同じ強さの電流がまたピッと流れることになります。2個のX線が全く同じタイミングで飛び込んで2倍の電流が流れる、というようなことは滅多に起こりませんから、電流パルスの高さを測りさえすれば、個々のX線のエネルギー、つまり波長がわかるのです。一方X線の強度の方は、単位時間に発生するパルスの数を数えればわかりますから、特定の波長のX線がどのくらいの強さで来ているかが測定できるわけです。図7の例で言えば、青で示した波長の短いX線が飛び込むと高いパルスが、赤で示した波長の長いX線が飛び込むと低いパルスが発生します(緑はその中間)。そして青と緑と赤のX線はそれぞれ3個、1個、2個飛び込んでいますから、それぞれのパルスの数も3個、1個、2個です(実際にはパルスの数はもっと多いです)。このデータを、横軸にX線のエネルギー(つまりパルスの高さ)、縦軸に強度(つまりパルスの数)をとって表示すれば、X線分光の完成です。このようにしてX線を分光する方法を、エネルギーの違いを検出するという意味で、エネルギー分散分光(
Energy
Dispersive
X-ray Spectroscopy = EDX または EDS)と呼びます(この方式の検出器のことをEDXと呼ぶ場合もあります)。
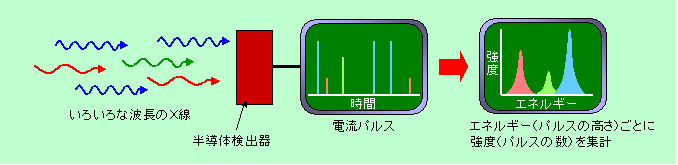
図7 EDXによるX線分光
EDXのいいところは、何と言っても小型、簡便という点です。何せ、小さな半導体素子に電圧をかけて電流を測るだけですから。その特徴を活かして、電子顕微鏡の付属装置としても普及しています。電子線の照射は専門の電子顕微鏡に任せて、出て来るX線の分析だけを担当しようというわけです。
EDXの弱点は、分解能がイマイチだということです。検出器の性格上、たとえ同じエネルギーのX線が来ても、必ずしもピッタリ同じ電流パルスの値にはならず、多少のバラつきが出てしまうのです。そのため、例えばある元素からのX線によるパルスが少し高めに出て、別の元素の領域に入ってしまう、ということも起こります(図7右の例では、青と緑の領域がちょっと重なっています)。こうなると、本当はないはずの元素があるように見えたり、エネルギーが近いX線を出す元素どうしの見分けがつかなかったり、ということが起こってしまうのです。この弱点を克服できるのが、もう一つの分光方法、波長分散分光(
Wavelength
Dispersive
X-ray Spectroscopy = WDX または WDS)です。
先に、X線は波長が短すぎて普通の回折格子は使えないと書きました。それでは「普通でない」回折格子、格子の間隔がものすごく狭い回折格子ならば分光できるのではないか? その通りです。ではどのくらい格子間隔が狭ければ使えるかと言うと、分析するX線の波長ぐらいですから、nmレベル、ということになります。こんな狭い間隔で格子を刻むことなどとても不可能・・・・なのですが、自然界にはそれがちゃんと用意されています。そうです。結晶を使うのです。
X線回折では波長のわかっているX線を使って結晶の格子間隔を求めていましたが、その逆に、格子間隔のわかっている結晶を使って、X線の波長を求めるわけです。
図8はWDSの概要を示したものです。試料から出て来たX線を分光用の結晶に入れ、回折して来るX線を検出器で捕まえます。波長が長いほど大きな角度で回折しますから、これによって波長ごとのX線強度が求められるのです。いろいろな元素から出るX線の波長は0.01nm〜数nmの範囲にわたっており、一種類の結晶で全部をカバーするのは難しいので、普通は格子間隔の違う数種類の分光結晶を使い分けています。
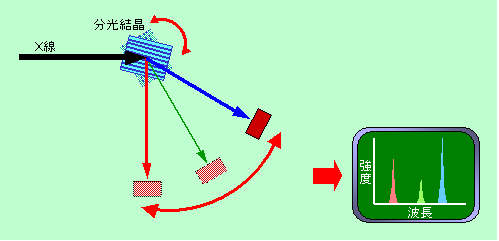
図8 WDXによるX線分光法
WDXはEDXと比べて分解能が高いのが特徴で、X線波長がちょっとだけ違う元素もきっちり見分けられます。その反面、装置がどうしても大きくなりますから、SEMなどに付属させるというわけには行かず、精密な分析を目的としたEPMA専用機や、高級な蛍光X線分析装置に搭載されています。このような使い分けがあるので、一般的にはEPMAと言えば検出器はWDX、というのが常識になっています。ただし原理的に考えれば、検出器としてEDXを搭載したEPMAというのがあってもおかしくはありません(SEMにEDXを付属させた装置などは、まさにこれです)。また、よくEDXとEPMAを対比した話を耳にしますが、厳密に言えばEDXに対比されるのはWDXであって、EPMAではないのです。
電子を検出する分析法
電子を叩き出した後の空席に別の電子が落ち込む時に出るX線を対象にするのがX線分光でしたが、叩き出された電子そのもの(光電子)も、当然、分析対象になり得ます。これが光電子分光です(光ではない電子に対して「分光」というのも妙な感じがしますが、エネルギーの違いを分析する手法である「Spectroscopy」または「Spectrometry」に「分光」という日本語を当ててしまったので仕方ありません)。刺激として与えるエネルギー(電子を叩き出すエネルギー)に紫外線を使う場合とX線を使う場合があり、それぞれ、紫外線光電子分光(Ultraviolet Photoelectron Spectroscopy = UPS)、X線光電子分光(X-ray Photoelectron Spectroscopy = XPS)と呼ばれます。なお、XPSはESCA(Electron Spectroscopy for Chemical Analysis)とも呼ばれます。物理系の人はXPS、化学系の人はESCAという呼び方をすることが多いようです。X線はそこそこ奥まで入り込みますが、分析対象となる光電子の方がごく浅いところからしか脱出できませんので、UPSもXPSも表面から数nmレベルの情報だけを取り出す最表面分析の手法です。
図6の説明では、単に電子のエネルギーが高い、低い、という言い方をしましたが、ここではもう少し詳しく電子のエネルギーを見ておきましょう。マイナスの電荷を持った電子はプラスの電荷を持った中心の原子核に引っ張られ、束縛されています。エネルギーで言うと、外を自由に飛び回る状態と比べてエネルギーが低く安定になっており、ちょうど井戸の中に収まっているような状態です。原子核に近いほどエネルギーは低く、井戸の奥深くにいることになります。ここに紫外線やX線を当てると、図9に示すように、そのエネルギーをもらった電子が井戸の外に飛び出すのです。エネルギーの大きいX線は井戸の深いところの電子も叩き出すことができますが、エネルギーの小さい紫外線はもっぱら井戸の浅いところの電子を叩き出すことになります。
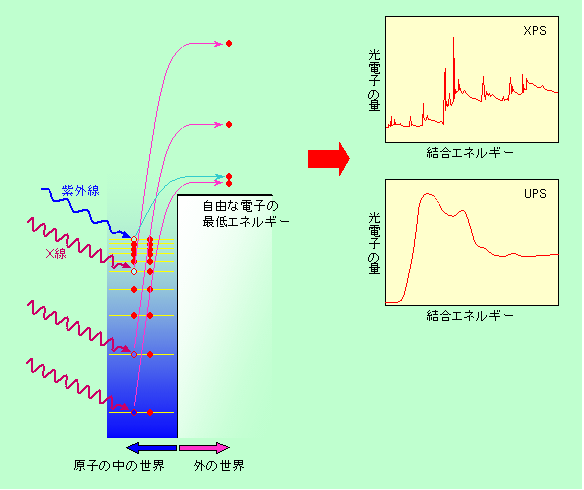
図9 叩き出された電子を直接分析する光電子分光
外に出た光電子は、初めの紫外線やX線のエネルギーから、井戸を登るのに使った分を差し引いた残りのエネルギーを持っています。井戸の深いところから飛び出した光電子ほど多くのエネルギーを消費していますので、残ったエネルギーは小さく、ノロノロと飛ぶことになります。ということは、逆に飛び出した光電子のエネルギーを測ってやれば、その電子が元々どのくらいの深さにいたのかがわかるということを意味します。つまり、光電子のエネルギー分布は、そのまま井戸の中の電子のエネルギー分布に対応するのです。井戸の深いところと浅いところでは電子の分布状態はかなり違っていて、深いところではエネルギーが飛び飛びになっていますが、浅いところでは隣近所の原子どうしで混じり合ってエネルギーの間隔が狭く(ほとんど連続に)なっています。そのため、図にも示しているように、XPSでは飛び飛びのエネルギー分布が、またUPSでは連続的なエネルギー分布が見られるのが普通です。XPSで観測される飛び飛びのエネルギー分布は原子中の電子状態そのものですから原子の種類を特定するのに有効です(多少は周りの影響も受けるので、原子の状態分析にも使えます)。一方UPSは、原子と言うよりも原子が集まった物質の電子の出し易さ(電子を出すのに必要なエネルギーを「仕事関数」と言います)を調べるのに使われます。
(注):XPSやUPSでは測定結果を表示するのに光電子のエネルギーをそのまま使わないで、初めにぶつけたエネルギーから飛び出した光電子のエネルギーを差し引いた値で表示するのが普通です。これは井戸を登るのに消費したエネルギー、つまり元の電子が収まっていた深さに対応し、電子が原子核に引き付けられていた程度を表すという意味で「結合エネルギー」と呼ばれています。
光電子分光の装置は、およそ図10のような形をしています。目立つのは半球形の、お椀を伏せたような形の部分で、これが静電半球と呼ばれる光電子のエネルギーを分析する道具です。
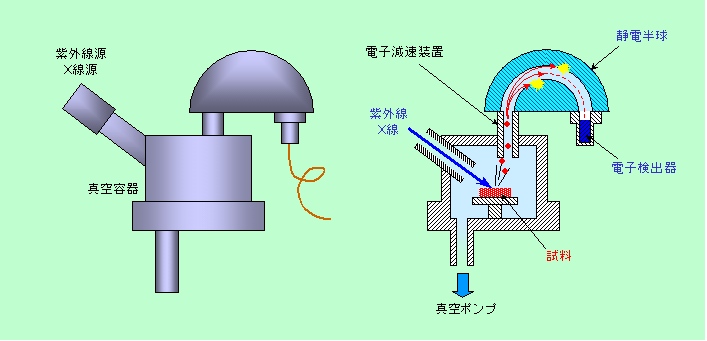
図10 光電子分光の装置
X線と違って電子線は真空中でないとまともに飛びません。特に光電子分光では電子のエネルギー、つまり速さを問題にしますから、空気の分子にぶつかって乱されるようなことがあっては困ります。そこで、装置の中はかなりハイレベルの真空状態になっており、その中に置いた試料に紫外線やX線をぶつけ、飛び出して来る光電子を静電半球に導いて分析するようになっています。静電半球は大きさの違う2つのお椀を重ねたような構造で、内側がプラス、外側がマイナスになるように電圧がかかっています。ここに入って来た光電子は電場に引っ張られて進路が曲がるのですが、エネルギーの大きい(速い)光電子は曲がりきれずに外側の壁にぶつかり、エネルギーの小さい(遅い)光電子は曲がりすぎて内側の壁にぶつかりますから、ちょうどよいエネルギーを持った電子だけが静電半球を通り抜けて、反対側の検出器まで届きます。このようにして、特定のエネルギーを持った光電子だけを選び出すことができるのです。
いろいろなエネルギーを持った光電子を測定するには、静電半球にかける電圧を少しずつ変えて測って行けばよいのですが、実際には、静電半球の電圧は固定し、代わりに入口に設けた装置で光電子の速度を落とす方法が一般的です。どの程度減速すれば光電子が静電半球を通り抜けるようになるか、ということを調べて、元の光電子のエネルギーを判定するわけです。
XPSなどで電子が叩き出された後にできた空席はどうなるでしょうか。当然、図6の蛍光X線で説明したように、高いところの電子が空席を埋めるために落ちて来ます。この時に余ったエネルギーをX線として放出するのが蛍光X線でした。ところが、X線が出る代わりに、余ったエネルギーをもらって別の電子が飛び出す場合があます。これを、発見者の名前を取って「オージエ電子」と呼びます(図11)。
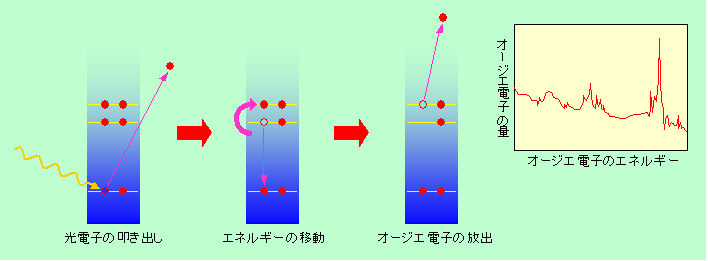
図11 オージエ電子の放出
放出の仕組みからわかるように、オージエ電子のエネルギーは初めのX線のエネルギーとは関係ありません。完全に原子内部の電子のエネルギー状態だけで決まります。ですから、このオージエ電子を分析すれば、原子の種類やその状態が特定できるのです。これがオージエ電子分光(
Auger
Electron
Spectroscopy = AES)です。外から来た強いX線などで直接に叩き出された電子と違って、オージエ電子のエネルギー源は同じ原子内での電子の飛び降りですから、XPSで捕まる電子のエネルギーと比べてオージエ電子のエネルギーは小さめです。実際にXPSの測定結果をよく見てみると、エネルギーの低い領域(先のXPS的な表示方法で言えば、結合エネルギーの大きい領域)にオージエ電子を見つけることができる場合があります。つまり、XPSの装置でオージエ電子も観測できる、ということです。それならばXPSの延長で済ませてしまえばよさそうですが、実はAESには非常に大きな利点があります。それは、初めの刺激としてX線ではなく電子線も使うことができる、ということです。普通の光電子分光で紫外線やX線の代わりに電子線を使おうとすると、叩き出された電子と、初めにぶつけた電子の跳ね返りとが混ざってしまい、分析ができなくなってしまいます。ところがオージエ電子はエネルギーが小さいので、ぶつけた電子線と重なることなく分析ができるのです。
それでは、X線ではなく電子線を刺激に使う利点は何でしょうか。これはちょうど、蛍光X線分析とEPMAとの違いに相当します。つまり、電子線はX線と比べて非常に小さくビームを絞ることができ、0.1mmがせいぜいのXPSと比べて2桁小さいμmレベル、さらに特殊な装置ならばもう1桁小さい領域にまで絞った分析が可能なのです。これを活かして、半導体の回路などの微細な構造体の分析などに活躍しています。
電子を検出する分析法としては電子顕微鏡(TEM,SEM)もありますが、これらについては
電子顕微鏡の話や
SEMの話で詳細に説明していますので、ここでは省略します。ちょっと変わったところでは、試料にイオンをぶつけて、そこから出てくる二次電子を捕まえる走査イオン顕微鏡(
Scanning
Ion
Microscope = SIM)というのもあります。大きなイオンは電子のようには試料の中に侵入できませんから、表面の原子一層分ぐらいの情報が得られることになります。普通は独立した装置ではなくて、
電子顕微鏡の試料作りに使われる収束イオンビーム装置に二次電子検出器を付属させた形で使われます。
イオンを検出する分析法
分析法の紹介の最後は、イオンを検出する方法です。仕組みは単純で、試料に酸素やアルゴン、セシウムなどのイオンをぶつけて、試料表面から叩き出される物質のうちイオン化しているもの(二次イオン)を検出し、試料の組成を調べるのです。ちょうど
走査型電子顕微鏡(SEM)の電子をイオンに置き換えた形ですね。二次イオンの分析には質量分析法が使われますので、「二次イオン質量分析(
Secondary
Ion
Mass
Spectroscopy = SIMS)」と呼ばれます。質量分析にはいくつかの方法がありますが、代表的なのは、XPSの静電半球と同じように、飛んで来たイオンに電場や磁場をかけて進路を曲げ、条件に合ったイオンだけを通過させる、というものです。電場や磁場から加わる力はイオンの電荷によって決まりますから、電荷の大きさが同じならば重い方が曲がりにくい、ということを利用して、イオンを重さごとに分けて分析するのです。
SIMSでは刺激も応答も、電子と比べてはるかに大きなイオンですから、本当に一番表面にある原子しか関係しない、最表面分析法です。原子から副次的に発生する電子やX線ではなくて、原子そのものを直接に捕まえますから、感度が非常に高いのが特徴で、他の表面分析法で検出できる限界の濃度はせいぜい0.1%というところですが、SIMSではppm〜ppbレベルの微量成分も検出できるのです。また、光電子分光などが苦手とする水素やリチウムなどの軽い元素も分析できますし、分析領域も、オージエ電子分光には及びませんが数μm程度に絞れるなど、いろいろな特徴を持っています(図12)。
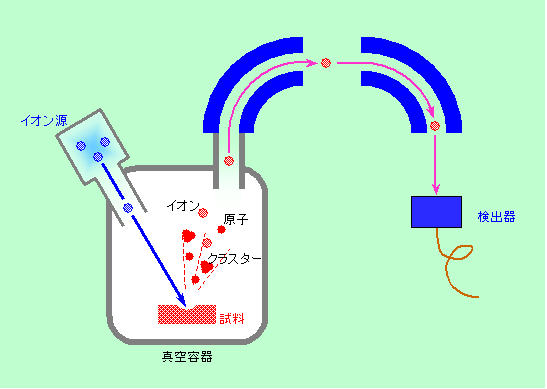
図12 イオンでイオンを叩き出す SIMS
このように表面分析法としていいことずくめのようなSIMSですが、欠点は表面を破壊してしまうことです。表面の原子そのものを叩き出しているのですから、これは仕方ありません。ただ、これを逆に利用して試料を掘り進みながら分析して行くことができます。時間と共にどんどん深いところの情報が出て来るようになりますから、試料の組成を立体的に見ることができるのです。(他の分析方法でも、イオンをぶつけて試料を掘りながら分析できるようになっている装置もあります)
不感時間という妙なもの
電磁波にしろ電子にしろイオンにしろ、これらのエネルギーの粒は最後は何らかの検出器に受けて量を測る必要があります。検出方式にはいろいろなものがありますが、基本的にはEDXのところで説明したように、エネルギーの粒が飛び込むたびに信号がピッ、ピッと出るようになっています。この時、検出器には何らかの変化が起こっているのですが(だからこそ信号が出るのです)、次々に飛び込んでくる粒を検出するには、そのたびにちゃんと元の状態に戻っていなければなりません。でないと変化が蓄積して、同じ感度で検出を続けることができなくなってしまいます。ところが実際には、検出器の状態は一瞬で元に戻るわけではありません。元の状態に戻るには一定の時間が必要で、その間は粒が飛んで来ても反応できない「死んだ」状態になるのです。この時間を「不感時間(Dead Time)」と呼びます。ちゃんと検出するには、不感時間などはない方がいいように思えます。それなのにEDXのマニュアルなどには、たいてい「不感時間が20〜30%になるように調節しなさい」というようなことが書いてあります。わざわざ2割、3割の時間は検出不能の「死んだ」状態にしろとは、少々妙な話ですね。これはどういうことでしょうか。
エネルギーの粒が一個飛び込んで信号が出た後、検出器が元の状態に回復するまでに0.01秒かかるとしましょう。1秒間に40個の粒が飛び込むような条件で測定した場合、不感時間のトータルは0.4秒(つまり不感時間40%)ですから、この検出器が実際に活動しているのは残りの0.6秒だけです。単純計算すると、この状況で検出できる粒は、検出器が生きている0.6秒の間に飛び込んで来た24個だけで、残りの16個は数え損ねてしまう、ということになります。次に(例えばEPMAの照射電子線を弱くするなどして)1秒間に検出器に飛び込む粒の数を20個に減らしてみましょう。そうすると確かに不感時間は0.2秒に減るのですが、粒の数が全部で20個しかありませんから、検出できる粒はその8割で16個となり、装置の感度としては落ちてしまいます。それでは逆に検出器に飛び込む粒の数を毎秒80個まで増やすとどうなるでしょうか。今度は不感時間が0.8秒もありますから、結局は全体の8割を数え損ねてしまい、やはり16個しか検出できないのです。この様子を図13にグラフで表しました。(なお、ここでは飛び込んだ全ての粒が全く同じ不感時間を発生させるという単純なモデルで考えました。この他の考え方として、不感時間中に飛び込んだ粒は新たな不感時間を発生させないとするモデルや、その中間のモデルなどいろいろあり、グラフの形も少し違って来ます。どのモデルが実際とよく合うかは、その時々の状況によります。)
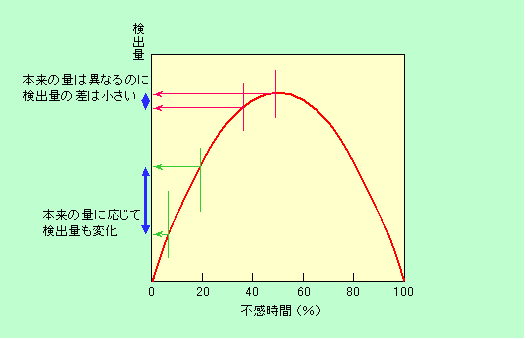
図13 検出されるエネルギー粒子の数と不感時間との関係
左の方では粒の絶対数が少ないことが、また右の方では不感時間が長くなって数え損ねが増えたことが影響して、検出される粒の数が減ってしまっています。真中あたりの条件の時に、最も多くの粒が観測できるのです。それでは、いつもこの真中あたりの条件で測ればよいかというと、そうでもない、というのがややこしいところです。その理由はこうです。
真中附近は検出される粒が多くて、確かに検出感度は最大です。しかし、全体の半分ぐらいは不感時間ですから、実際に検出器に飛び込んで来ている粒の半分程度しか検出できていません。つまり、量に関する情報がウソになるのです(もちろん、さらに右側の領域ではもっと大ウソになります)。また真中附近はグラフの傾斜がなだらかなので、検出器に飛び込む粒の数が少々変化しても(言い換えれば不感時間が少々変化しても)、実際に検出される粒の数はほとんど変わらない、という問題も発生します。要するに、量の変化に鈍感なのです。これに対して、グラフの左の方では、飛び込む粒子数、つまり不感時間の変化に応じて検出される粒子数が大きく変わりますから、量の変化を敏感に感じ取ることができます。最も量の変化に敏感なのは、言うまでもなく、グラフの傾斜が最も急な左端の点、ということになります。
以上のように、真中附近の条件は、或る成分があるかどうかだけを感度を目一杯上げて判定するような時には都合がよいのですが、量についての情報も欲しい場合(普通はそうです)には具合が悪いのです。かといって左端の条件ではあまりに感度が低すぎますから、感度もそこそこ、量を測る目的もそこそこ果たせる、不感時間20〜40%あたりがよく使われるのです。(検出器自体の感度が非常に高く、精密に量を測る必要がある場合には、不感時間がほとんどない左端に近い条件も使われます。)
表面分析のポイント
これまで表面分析の方法についていろいろと書いてきましたが、表面分析に限らず、分析を成功させるポイントは、「何を調べたいのか」をはっきりさせた上で、最適な分析手段を選ぶことにある、と私は思っています。分析対象はおよそどんなものなのか、その組成を調べたいのか、構造を調べたいのか、表面からどのくらいの深さまでの情報が知りたいのか、といったことをできるだけはっきりさせ、その他の条件(試料の大きさは?、破壊してもよいのか非破壊で調べなければならないのか?、真空にしても大丈夫か?、などなど)を考慮して、どんな方法で分析するかを決めるのです。これが正確にできたらほとんど成功したようなもので、後はお金さえ出せば(一種類の測定だけでは不十分かもしれませんが)とりあえずは望みの結果が得られるはずです。分析の達人というのは、この手法の選択が適切にできる人なのだと思います。
雑科学ホーム
hr-inoueホーム