
雑科学ホーム
hr-inoueホーム
● クロマトグラフィーの話 ●
混ざった物を分ける技術
ここに何種類かの液体が混ざったものがあるとします。この中から欲しい成分だけを取り出すには、どのようにしたらよいでしょうか。これにはいろいろな方法が考えられます。例えば、その成分が蒸発しやすいものならば、加熱して初めに蒸発して来る部分を集めればよいでしょう(蒸留)。あるいは、その成分だけが特によく混ざる別の液体があるならば、その液体と一緒に振り混ぜて、目的の成分だけを引っ張り出してやる、ということもできます(抽出)。とにかく、元の混合液体の中から別の場所(気体の中とか別の液体の中とか)に、目的の成分を引き出してやればよいのです。とは言っても、一発で狙った成分だけが取り出せることはめったにありません。他の成分もいくらかは混ざって来るのが普通です。そこで、一回取り出したものに対してさらに同じ操作を繰り返して、どんどん純度を上げて行く、という方法が採られます。これを連続的にできるようにしたのが「クロマトグラフィー」です。
「クロマトグラフィー(Chromatography)」というのは、元々は植物の色素を分離するために考え出された方法だそうで、「クロマト(Chromato)」というのは、「色」を意味するギリシャ語の「Chroma」から来ています。おそらく、多くの方が最初に接するクロマトグラフィーは、学校の理科の時間に出て来る「ペーパークロマトグラフィー」ではないかと思いますが、ここでは濾紙を使ってインクの色成分を分離する実験をしたのではないでしょうか。正に「色を分離する」クロマトグラフィーの原点ですね。しかし今では、クロマトグラフィーは大きく発展し、色素だけでなく、様々な化学成分を細かく分離して取り出したり、不純物を取り除いて精製したり、微量に含まれる成分を分析したりする手法として大活躍しています。この項では、こうしたクロマトグラフィーの基本的なところを見て行くことにしましょう。
基本は2つの「相」への分配
先ほど、「目的の成分を別の場所に引き出す操作を連続的に行うのがクロマトグラフィー」だと書きました。このことをもう少し詳しく見てみることにします。まず「別の場所」とは何か、ということです。例えば身の回りの空気を考えてみると、これは(ちょっとした気圧の差とか、車の排ガスが多い場所があるとかいう話は無視して)どこでも同じ成分、同じ性質を持っていると考えられます。バケツに入った水も、どこを取っても同じ成分、同じ性質ですし、海の水も、塩を初めとするいろいろなものが溶け込んではいますが、やはり成分・性質は均一、と言えます。このような、成分・性質がどこを取っても同じである状態を「相」と言い、空気のような気体ならば「気相」、水のような液体ならば「液相」と呼ばれます。一つの相の中は均質ですから、どこにも境界はありません。これに対して、空気と水とでは明らかに成分も性質も違い、はっきりした境界がありますから、それぞれ別の相です。また水の中に氷が浮かんでいる状態も、成分こそ同じですが、水と氷では性質は全く違っており、誰の目にも明らかな境界があります。つまり氷水は一つの相ではなく、液相である水と、固相である氷に分かれた状態、ということになります。ここまで来ればもうおわかりと思いますが、先ほどの「別の場所」というのは、正にこのような「別の相」、「相が異なる場所」という意味なのです。ついでに言うと、液体でも気体でもなくなった
超臨界状態も、「超臨界相」と呼ばれる、境界のない一つの相です。
それでは「目的の成分を別の相に引き出す」とは、どういうことでしょうか。実はこれは、「引き出す」のではなく「勝手に移って来る」というのが正確な言い方です。2つの相が接していて、第3の成分である目的物質がどちらでも好きな方に行ける状態にあるとします。この時、どちらにどれだけ分配されるかは、成分の種類と相の種類・状態によって勝手に決まってしまうのです。例えば、密閉された容器の中に水と空気が入っており、水中には水よりも蒸発しやすいアルコールが溶け込んでいるとします。そうすると、水の中のアルコールは徐々に蒸発して空気中に出て行き、空気中のアルコール濃度と水中のアルコール濃度がある決まった値になったところで安定します(実際には蒸発が止まるのではなく、蒸発する速度と水中に戻る速度がつり合った状態になっています。いわゆる平衡状態です)。その時の空気と水へのアルコールの分配比率は、その場の温度や圧力によって決まってしまい、自由に変えることはできません。このような2つの相への分配が、クロマトグラフィーの基本になります。相の組み合わせとしては、固相どうしだときちんと接触しませんし、気相どうしだと完全に混ざってしまいますから具合が悪いですが、その他の組み合わせはいろいろと考えられます。固相/液相、固相/気相、液相/液相(ただし2つに分離することが必要)、液相/気相、いずれもOK、さらには
超臨界相も使うことができます。
このような2つの相に第3の成分が分配されるのには、図1に示すようにいくつかのタイプがあります。一つ目は、互いに混ざり合わない2つの液相に第3成分が溶け込む場合のように、文字通り「分配」されるものです(図1(a))。この場合、第3成分はそれぞれの相に完全に溶け込んだ形になり、どちらの相に溶け込みやすいかで分配比率が決まります。二つ目は、相の一方が固体の場合に見られる型で、第3成分は普通は固体の内部には溶け込みませんから、表面への「
吸着」、という形になります(図1(b))。冷蔵庫脱臭剤の多くには表面積の大きな活性炭が使われていますが、この活性炭の表面に臭いの元が吸着されるようなケースがこれに当たります。臭いの元となる分子が、空気中(気相)と活性炭表面(固相)とに分配されるわけです。もちろん優秀な脱臭剤ほど、固相への分配比率が高くなりますが、全部が固相に移って空気中の濃度がゼロになる、ということはありません。平衡状態になるのですから、どんなに偏るとしても、必ず両方の相に取り分はあるのです。
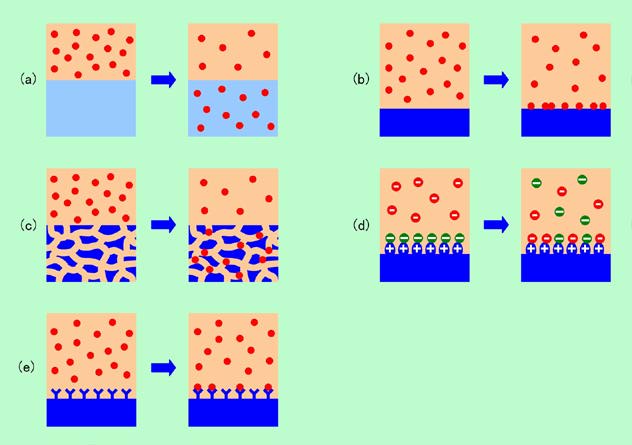
図1 2つの相に第3成分を分配する方式いろいろ
三つ目は、一方の固体が小さな孔をたくさん持った構造になっているケースです。その孔の中に第3成分が入って行くのですが、手ごろな大きさの孔がたくさんあるほど多くの第3成分が侵入することになります(図1(c))。この場合、第3成分が固体の中に溶け込むわけではありませんが、分子レベルの孔に入って行くのですから、表面に吸着するのとも違っていて、両者の中間のような感じです。実際には孔の内壁と何らかの作用をすることもあるので、「吸着」と厳密に区別することができない場合もありますが、後で出て来るようにクロマトグラフィーの手法として非常に特徴的ですので、別に考えておいた方がよいでしょう。
この他に、吸着の一種と言えなくもありませんが、特別な作用でもって分配を起こさせるケースがあります。その一つが図1(d)の「イオン交換」という現象です。固体の表面にイオンに解離する性質を持った部分があり、ここにあるイオンと交換することで、液相中にある第3成分のイオンを表面に取り込むのです。また、特定の物質とだけくっ付く性質を一方の相に持たせてやれば、その物質だけを選んで取り込むこともできます(図1(e))。生物の抗原・抗体反応などがよい例で、固体表面に抗体を植え付けておけば、特定の抗原だけを捕まえることができるのです。
分配を連続的に行う
目的の成分を分離するために2つの相への分配を利用するわけですが、1回の分配だけできれいに分けられるとは限りません。その場合は、分配操作を何回も繰り返す必要があります。この繰り返しの様子を、「蒸留」を例にとって見てみましょう。言うまでもなく「蒸留」というのは、「複数の成分が混ざった液体を加熱して目的の成分を蒸発させ、蒸気を冷やして目的成分を回収する」操作ですね。先ほどの言い方に沿えば、「目的成分を液相から気相の方へ引き出して分離する」、ということになります。この時、気相には目的成分だけが出て来ているとは限りません。と言うよりもむしろ、他の成分も混ざっているのが普通でしょう。そこで分離のレベルを高めるために、図2(a)のような多段階の蒸留が行われます。
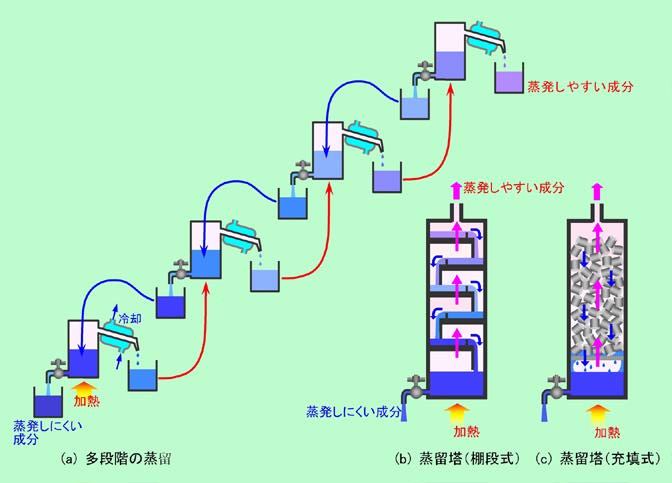
図2 蒸留の分離精度を高める
左端が1段目の蒸留です。気相を冷やして回収した液には(蒸発しやすい)目的成分が多く含まれているはずですが、他の成分もある程度は一緒に出て来ています。そこで、この液を(赤矢印のように)第2の容器に移して2段目の蒸留をします。この時、前と同じ温度にすると全部が蒸発してしまいますから、前よりも少し低い温度にする必要があります。ここで回収される蒸留物は、元々濃度が高い液からスタートしているのですから、1段目よりも目的成分を多く含んでいるはずです。これをさらに第3の容器に移して3段目の蒸留をすれば、目的成分の濃度はさらに高くなります。このようにして次々に蒸留を重ねて行くことで、純粋な目的成分に近付いて行くのです。一方、各段階で容器に残った液にも目的成分は残っていますから、これを捨ててしまうのはもったいないので、(青矢印のように)一段前の容器に戻して再度蒸留することもできます。この残った液を前段に戻すという操作は、裏を返せば、蒸発しにくい成分を前の方(図では左の方)に集める、ということでもあります。つまり、右端からは蒸発しやすい成分が取り出され、左端には蒸発しにくい成分が残ることになるのです。
このような多段階の蒸留操作を連続的にできるようにしたのが、図2(b)や図2(c)の蒸留塔です。図2(b)は棚段式と呼ばれる方式で、途中に何段もの棚が設けてあり、それぞれの棚で気相/液相間での成分の分配が起こります。つまり、それぞれの段が1個の蒸留装置になっているのです。温度が上がって軽くなった気相は棚に設けられた孔(図では省略)を通って一つ上の段に上がって行き、一方、棚に溜まった液相は棚の縁の壁を乗り越えて、重力で下の段に降りて行きます。図2(a)の多段蒸留の装置と比べると、気相をいちいち液化してから次に移すのではなく、気体のまま次の段に送るようになっている点が違っていますが、この点を除けば全く同じ仕組みになっていることがわかると思います。
図2(c)は、このような段も省略して、代わりに円筒型や鞍型の充填物を詰め込んだ「充填式」の蒸留塔です。この方式では、充填物の表面に付着した液体が「分配」の場になります。蒸発しやすい成分を多く含んだ気相が上に向かい、蒸発しにくい成分を多く含んだ液相が下に下りて行く様子は、棚段式と変わりありません。いずれにしても、図2(a)のような逐次方式(バッチ方式と言います)と比べて図2(b)や図2(c)の連続方式の方が操作が簡単で効率的、と言えるでしょう。
以上は連続的に「蒸留」する話でしたが、次に「抽出」について見てみましょう。今度は両方とも液相ですが、基本的には蒸留の場合とよく似ています。
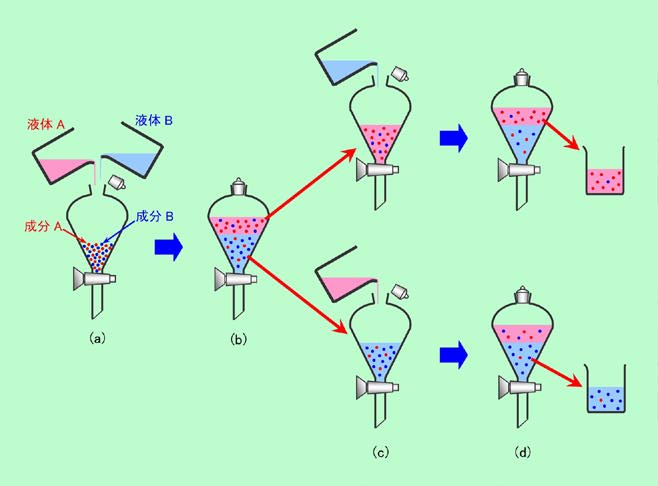
図3 分液ロートで抽出を繰り返す
図3はバッチ式の抽出操作を表しています。初め、左端の(a)ように分液ロートに赤丸で示した成分Aと青丸で示した成分Bとが入っているとします(分液ロートというのは層分離した液を分けて取り出すのに便利な器具で、図のように下にコックが付いており、そのコックを開けて下側の液だけを取り出せるようになっています)。ここに互いに混ざり合わない、赤色で表した液体Aと青色で表した液体Bを注いでよく溶かし、静かに置いておくと、やがて(b)のように2層に分離します。この時、成分Aは液体Aによく溶け、成分Bは液体Bによく溶けるとします。ここでは成分Aが液体Aと液体Bに分配される比率は3対1、成分Bが液体Bと液体Aに分配される比率も同じく3対1であるとしましょう。すると、16個の丸で示された成分Aのうち12個は液体Aに、残りの4個は液体Bに分配されることになります(成分Bはもちろんこの逆)。成分Aが液体Aに、成分Bが液体Bに優先的に抽出されたわけですね。しかし余分な成分がそれぞれ4個も混じっているということで、純度としては75%しかありませんから、もっと純度を上げるために2回目の抽出を行ないます。(c)のように液体Aと液体Bとを分離し、それぞれに相方の液体を入れてもう一度混ぜるのです。その結果、(d)の上段に示したように、液体Aの中には成分Aが9個に対して成分Bは1個だけ、下段の液体Bの中には、逆に成分Bが9個に対して成分Aは1個だけになりました。それぞれ純度が90%まで上がったわけです。上段の液体Bや下段の液体Aには成分Aと成分Bとが半分ずつ入っていますが、これも捨ててしまう必要はありません。それぞれを取り出してさらに抽出操作を繰り返せば、同じように純度を高めることができます。
分液ロートを使うのではなくて、仕切りで細かく分かれた小部屋の中で抽出するようにしたのが図4(a)です。形としては、図2(b)の棚段式蒸留塔に近いと言えますね。蒸留塔では気相と液相とが互いに逆方向に動いていましたが、ここでは下側の液体B(青色)は動かず、上側の液体A(赤色)だけを右に一区画ずつ送って行きます。要は2つの相が相対的に動いて相手を替えて行けばいいのであって、液体Aだけを動かしても、液体Bだけを動かしても、あるいは(蒸留塔のように)両方を動かしても構わないのです。
この図でも、成分A(赤丸)が液体Aと液体Bに分配される比率は3対1、成分B(青丸)が液体Bと液体Aに分配される比率も3対1、としました。ただし、各成分をたった16個の丸で表していますから、後の方になるにつれて割り切れないケースが出てきます。その場合は3対1に近い整数にまるめて配分していますので、あまり正確な図にはなっていませんが、雰囲気は掴めると思います(もっとキッチリと計算した結果は後で出て来ます)。
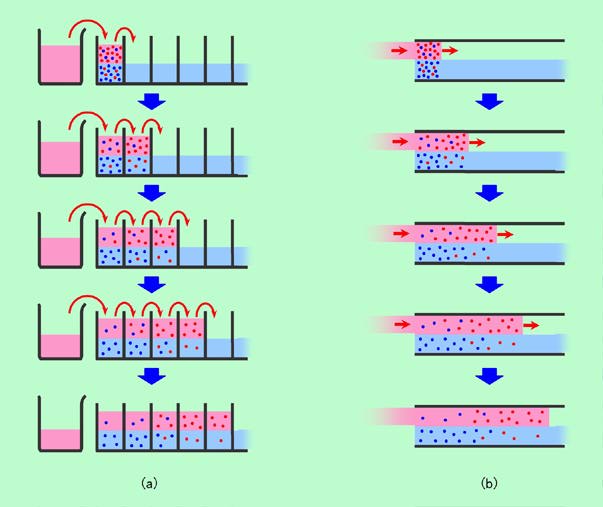
図4 連続で抽出する
図4(a)の一番上は1回目の分配の状態、2番目は液体Aを一区画右に送った2回目の分配の状態を表していますが、これらの様子は図3の分液ロートの場合と全く同じです。3番目以降はさらに抽出を進めた状態に相当し、成分Aは液体Aに溶け込んで右の区画へと移動して行くのに対して、成分Bは液体Bに引き止められて、あまり移動できません。厳密に言うと成分Bも少しずつ液体Aに溶けて右に移るのですが、その速度が成分Aよりも遅いのです。その結果、操作が進むにつれて成分Aはどんどん先に行き、取り残された成分Bと分離して来ます。一番下の5段階目では、既に右の2区画は成分Aのみ、左の2区画は成分Bのみになっていますが、もっと操作を進めれば、2つの成分は完全に分離してしまうのです。
このようにして連続的な抽出ができるわけですが、それならばいっそのこと、小部屋の仕切りもなくしてしまったらどうでしょうか。それを示したのが図4(b)で、管状の入れ物の下半分に液体Bを溜めておき、上半分に液体Aを流すようになっています。図4(a)では濃度の違う液が仕切りで完全に区切られていましたが、図4(b)の場合には仕切りがありませんから、横方向で多少混ざり合ってしまう可能性はあります。しかし管が十分に細く、液の流れが十分に速ければ、それほど混ざることなく液を移動させることは可能でしょう。そして、流れの途中のそれぞれの場所で成分Aと成分Bが2つの液に分配され、結局は図4(a)と同じように左右に分かれることになるのです。この図4(b)の方式が、まさしく本稿の主題である「クロマトグラフィー」で、下の動かない部分を「固定相」、上の移動して行く部分を「移動相」と呼びます。目的成分は、途中途中で固定相と移動相とに分配されながら進んで行き、移動相になじみやすい物質は速く、固定相になじみやすい物質は遅く移動することで、成分ごとに分かれて行くのです。
実際のクロマトグラフィー
クロマトグラフィーは基本的には図4(b)のようにして成分を分離する方法なのですが、実際には様々なタイプのクロマトグラフィーがあり、いろいろ種類分けがされています。例えば移動相の種類によって、ガスクロマトグラフィー(Gas Chromatography = GC)、液体クロマトグラフィー(Liquid Chromatography = LC)、超臨界流体クロマトグラフィー(Supercritical Fluid Chromatography = SFC)、などがあり、また固定相の状態によって、紙を使うペーパークロマトグラフィー(Paper Chromatography)や、粉末を薄く塗布したものを使う薄層クロマトグラフィー(Thin Layer Chromatography = TLC)、筒状の容器を使うカラムクロマトグラフィー(Column Chromatography)などに分けられます。さらに図1に示したような、成分が両相に分配される原理によって、分配クロマトグラフィー、吸着クロマトグラフィー、イオン交換クロマトグラフィーなどと呼ばれる種類があります。ですから正確に言おうとすれば、これらの特徴を全部並べて、例えば、「吸着」に基づく-「カラム」を使った-「液体」クロマトグラフィー、ということになるのです。なお、念のために言っておくと、「クロマトグラフィー(Chromatography)」というのは「手法」「技法」を指す言葉であり、「クロマトグラフ(Chromatograph)」はその「分析装置」のこと、そして「クロマトグラム(Chromatogram)」というのは、分析結果を示した図表のことです。ですから、「どこどこのメーカーのクロマトグラフを使って液体クロマトグラフィーで分析した結果、これこれのクロマトグラムが得られました。」ということになるわけです。
図5に代表的なクロマトグラフィーの装置(つまりクロマトグラフ)を示しました。
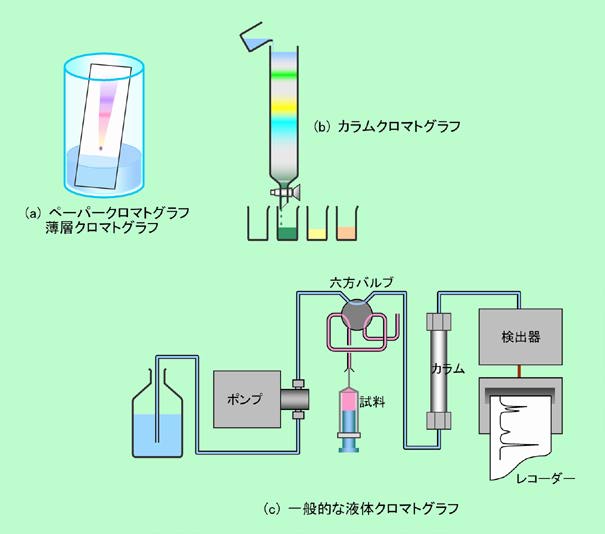
図5 実際のクロマトグラフの概観
図5(a)はペーパークロマトグラフィーや薄層クロマトグラフィーの例で、固定相が平面状なので「平面クロマトグラフィー」と呼ばれることもあります。ペーパークロマトグラフィーでは固定相は紙(濾紙)です。この紙の先端を適当な溶媒に浸すと、溶媒は毛細管現象で紙の繊維の隙間を通って上がって行き、これが移動相となります(普通は「展開溶媒」と呼ばれます)。紙の上に目的の試料をスポット状に付着させておくと、展開溶媒はやがて試料のところに到達し、これを溶かして上へと運んで行きます。試料の中の成分は紙への吸着と展開溶媒への溶解を繰り返しながら移動しますが、その時、展開溶媒に溶けやすい成分は速く移動し、紙に吸着しやすい成分は遅く移動しますので、成分ごとに分かれて来ることになるのです。つまりペーパークロマトグラフィーは、分配の原理で言えば吸着クロマトグラフィーであり、移動相の種類で言えば液体クロマトグラフィーであるわけです。薄層クロマトグラフィーも、紙の代わりにシリカゲルなどの粉末をガラス板などに薄く塗布したものを使うだけで、基本的にはペーパークロマトグラフィーと同じです。ただし厳密に言うと、シリカゲルなどの表面は裸ではなくて吸着した水で覆われており、試料はその吸着水の中に溶け込んでいる、と見なせる場合もあります。その場合はシリカゲルは固定相を保持する単なる土台(担体)に過ぎず、原理的にも吸着クロマトグラフィーではなくて、液相の固定相を用いた分配クロマトグラフィーであることになります。このように「吸着」と「分配」をはっきり区別するのが難しい場合も多く、一般的には両方の要素が含まれていると考えられます。
これらの平面クロマトグラフィーでは、色素のように成分に色が付いていればどこまで移動したかは一目瞭然ですが、無色の成分の場合には、紫外線を当てて蛍光を見るとか、別の試薬をかけて発色させるとかの工夫が必要になります。そして成分が移動した距離がわかれば、その距離を、展開溶媒の先端が進んだ距離で割った値(R
f値)で結果を表すことができます。固定相に保持されやすい成分ほどR
f値が小さいわけですね。
平面クロマトグラフィーは手軽で便利な方法ですが、成分を分離して取り出したり、量を正確に求めたり、微量成分を精密分析したりするのには向いていません。そのため、試料にどんな成分が含まれているかを大まかに調べたり、有機合成で目的の物質がちゃんとできているかをチェックする目的で使われることがほとんどです。また、次のカラムクロマトグラフィーを実施する前に、カラムに仕込むのと同じ充填剤を使って薄層クロマトグラフィーをやってみることで、どんな充填剤でどんな展開溶媒を使ったらよいかを事前に調べることもできます。
図5(b)はカラムを使った液体クロマトグラフィーの例です。ガラス等でできたの筒の中にシリカゲルなどの粉末を充填した「カラム」を用意し、上から展開溶媒を流します。もちろん、シリカゲルなどの充填剤が固定相で、展開溶媒が移動相になります(ここでも薄層クロマトグラフィーと同じように、充填剤の表面に吸着した水を固定相と考えた方がよい場合もあります)。普通はまずカラム全体に溶媒を通し、溶媒の液面が充填剤の一番上ギリギリのところまで来たら、試料を溶かした溶媒をそっと載せ、その液がまたギリギリのところまで入って行ったら、さらに溶媒を追加してどんどん連続的に流す、という方法が採られます。例によって固定相に保持されにくい成分ほど速く流下しますから、適当なタイミングで受けの容器を交換して行けば、出て来た成分を順に集めることができるのです。この例では展開溶媒は単に重力で降りて行くだけですから、成分が出るまでに普通は何時間もかかります。そこで上の部分を密閉して窒素などのガスで圧力をかけ、速く押し出せるようにした「フラッシュカラムクロマトグラフィー」と呼ばれる方法もあります。カラムクロマトグラフィーは、分析と言うよりも成分を分けて取り出すことに主な目的があり、特に不純物を除いて目的物を精製する用途に多く使われます。
カラムは長ければ長いほど、速い成分と遅い成分の差が大きく開きますから、分離の能力は高くなります。しかしそれだけ時間もかかるし溶媒も大量に必要ですので、むやみに長くすればよいというものではありません。通常は、長さは十数cmからせいぜい1mぐらいまでです。一方直径の方は、実験室レベルでは数mmで済む場合もありますが、精製したい試料の量が多ければ、それに応じて大きくした方が速く処理できます。実際に化学工場などでは直径1m以上のカラムも使われていて、長さよりも直径の方が大きい場合もあるのです。「カラム」というのは元々「柱」という意味ですが、こうなると樽か桶のような感じで、とても「柱」には見えません。
これらの実験用とか生産用のクロマトグラフィーとは別に、分析装置として発達したクロマトグラフィーがあります。図5(c)は液体クロマトグラフィーの例で、ポンプを使って、金属製の筒に充填剤を詰めたカラムに溶媒を送り込みます。ポンプが溶媒を送り出す圧力は数十気圧〜数百気圧にもなり、先のフラッシュカラムの比ではありません。それだけ短時間(数分〜数十分)にしっかり分離でき、高速の分析が可能、ということで、高速液体クロマトグラフィー(High Performance Liquid Chromatography = HPLC)と呼ばれています。分析試料はポンプとカラムの間に注入されます。試料を注入する部分の仕組みにはいろいろなものがありますが、ここで紹介しているのは六方バルブを使った方式です。図では試料は溶媒の流れに乗っていませんが、この状態から六方バルブを60°回転させると、溶媒が試料の入った管を通ってカラムに入るようになるのです。注入された試料は、カラムで成分ごとに分離した後に検出器に送られます。検出器のところでは、どんな種類の成分かはわからなくても、とりあえず移動相溶媒に含まれる成分の濃度がわかればよいので、屈折率や紫外線の吸収を測定する装置が取り付けられているのが普通です。測定された結果はレコーダーに送られ、濃度の変化がクロマトグラムとして記録されることになります。移動相溶媒に何も溶けていない時には屈折率や紫外線吸収は一定の値を示していますが、分離された成分が出始めると急に値が変化して、図のような成分の量に応じたピークが、時間の経過と共に次々に現れるのです。また、成分の量だけではなくて、その種類や分子構造についての情報も得られるように、質量分析計などの分析器を備えた装置もあります。
試料が注入されてから成分が検出されるまでの時間を「保持時間」と呼び、クロマトグラフィーでそれぞれの成分を特徴付ける要素となっています。試料に何が含まれているかが予測できる場合、その成分を事前にクロマトグラフィーにかけて保持時間を調べておき、実際の試料を分析した時の保持時間と比較することで、ある程度は確認することができるのです。
現在では、HPLCだけでなく多種多様の分析用クロマトグラフィーが使われています。以下に代表的なクロマトグラフィーをいくつか紹介しますが、基本形は図5(c)と大きく異なることはありません。
高速液体クロマトグラフィー(HPLC)
HPLCについては先ほど大雑把に説明しましたが、もう少し詳しく見てみましょう。まずカラムですが、一番単純なのは、普通のカラムクロマトグラフィーと同じようにシリカゲルやアルミナなどの充填剤を、内径3mm〜5mm、長さ10cm〜20cmの金属製の管に詰め込んだものです。これらの充填剤は、プラスとマイナスの電荷の偏りが大きい、つまり極性が大きい表面を持っています。このような表面には、同じく電荷の偏りが大きい、極性を持った物質がよく吸着し、極性の低い物質はあまり吸着しません(厳密に言うと、移動相溶媒も吸着するので、これとの競争になっています。
静電場の話、
吸着の話参照)。そこで、極性の低いヘキサンとかクロロホルムなどの溶媒を移動相として使うと、試料の中の極性の低い成分はあまり吸着せずに移動相に溶け込んで速く移動し、極性の高い成分はしっかり吸着して移動が遅くなって、きれいに分離されるのです(この場合も、シリカゲルなどの表面に吸着した水が固定相となり、水と移動相溶媒との間の分配で分離が進む、と見なせるケースもあります)。薄層クロマトグラフィーやカラムクロマトグラフィーと全く同じ状況ですね。それでは、移動相に水やメタノールなどの極性の高い溶媒を使ったらどうなるでしょうか。この場合、極性の高い成分は移動相にも固定相にも引っ張られて両相に同じように分配されますが、一方で極性の低い成分も、どちらの相からも嫌われて、結局同じように分配されます。つまり、状況が全く違うにもかかわらず、極性の高い成分も低い成分も分配の状態は同じようになってしまうのです。こうなると成分の分離はできません。きちんと成分を分離するためには、移動相と固定相は違う性質にしなければならないのです。
移動相と固定相の性質が違っていればよい、ということならば、さっきとは逆に移動相の極性を高く、固定相の極性を低くしてもクロマトグラフィーとしてちゃんと働くはずです。実際にこれは可能で、「逆相クロマトグラフィー」と呼ばれています(これに対して、先ほどのタイプを「順相クロマトグラフィー」と言います)。極性の低い分子は互いに引き合う力が弱いですから、集まって固体を作るよりも液体状態でいるものが多くなります。また固体を作ったとしても、他の物質を表面に引き付けて吸着させる能力はあまり高くはありません。そのため逆相クロマトグラフィーの固定相としては、普通は液体が使われます。しかし液体のままでカラムに仕込んだのでは簡単に押し出されてしまいますから、何らかの方法で固定しなければなりません。そこで、順相カラムでシリカゲル表面に水が吸着していたのと同じように、極性の低い物質をシリカゲルの表面に結合させて固定する方法が採られます。この場合、単なる吸着では水のように強固には固定できませんので、図6のような化学的な結合を利用します。図に示したのは最も代表的な炭素数18の分子を付けた例ですが、この他にもいろいろなタイプの逆相用カラムが作られています。
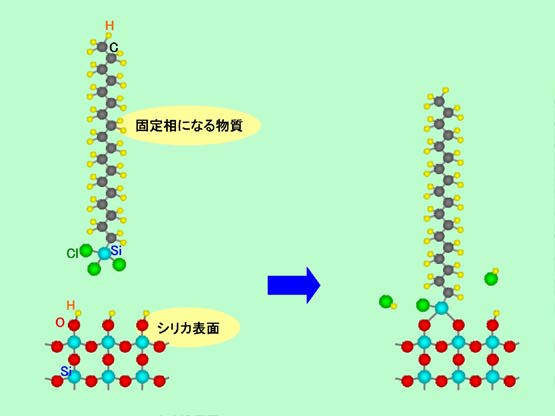
図6 極性の低い分子をシリカゲル表面に固定する
大半の逆相用の固定相はこのような「固体表面に固定された液相」なのですが、中には正真正銘の固体の固定相もあります。ポリマービーズと呼ばれる球状の樹脂などはその例です。また、ちょっと変わったところでは多孔性のグラファイトがあり、ベンゼン環(例の、亀の甲)を持った分子の分離などに使われています。
実はHPLCでは順相よりも逆相の方が多く使われます。その理由として、逆相で使う高極性溶媒の方が多くの種類の試料を溶かすことができるので、対応できる成分の種類が多いことや、順相では侵入した水の影響を受けやすいことなどがあります。とは言っても、順相でしか分析できないケースもありますから、逆相だけで済むというわけではありません。
極端に性質の違う成分が混ざっている場合には、一種類の移動相溶媒では足りない場合もあります。例えば逆相での分析で、高極性成分はすんなりと出て来たのに対して、低極性成分は固定相につかまっていつまで待っても出て来ない、ということも起こるのです。こんな時は、途中で溶媒の種類を変えて行くのが効果的です。初めは水だけ流しておいて、途中からメタノールやアセトニトリルを混ぜて行く、という具合です。これをグラジエント分析と言います。昔は手作業で切り替えなければならないのでいろいろとたいへんでしたが、今はコンピューターで複数のポンプを自由にコントロールできるようになっているので、条件さえ決めてしまえば後は簡単です(実は、条件を決めるのが一番難しいのですが・・・・)。
ガスクロマトグラフィー(GC)
図5(c)の溶媒の入ったビンとポンプの部分をガスボンベと圧力や流量を調整する弁に置き換えれば、ガスクロマトグラフィー(GC)の形になります。移動相はキャリアーガスと呼ばれる気体で、試料と余計な反応を起こさないように、主に窒素やヘリウムなどの不活性なガスが使われます。試料はキャリアーガスの流れに乗って移動するのですから、当然、気化する物でなければなりません(もちろん、試料を注入する部分を初め、装置の必要な部分をある程度加熱することは可能ですが、試料が変質してしまうほど高温にすることはできませんから、溶かしさえすればよいHPLCと比べると制約は多くなります)。気化した試料はカラムへと送られ、移動相と固定相とに分配されながら進んで行きます。この時、気体の移動相には、液体のように他の物質に積極的に働きかけて溶かし込む能力はほとんどありませんから、試料の移動相への分配はもっぱら試料自身の気化に頼っています。つまりHPLCと違って移動相の種類は分配にはほとんど関係なく、分配の状況は固定相の選択だけで決まってしまうわけです。移動相、固定相のそれぞれにしっかり溶け込んで行く液体クロマトグラフィーと比べて分離の性能はやや劣り、ピークが広がりやすい傾向がありますが、ガスの流れは速いので分析がスピーディーにできる、という特徴があります。
GC用の固定相も、固体のものと液体のものとがあります。固体の固定相としては、シリカゲルやアルミナ、
ゼオライト、活性炭などの吸着剤が使われ、これらを直径数mmのステンレス管やガラス管などに詰めてカラムにしています。長さはいろいろですが、普通は数十cm〜5mぐらいでしょう。ガスは液体よりも簡単に通り抜けるので、HPLCと比べて長いカラムになっています。HPLCに固体の固定相を使った場合は、試料と移動相溶媒が競争して吸着するのですが、GCの場合にはキャリアーガスの吸着はほとんどないので、試料の成分だけが吸着したり気化したりを繰り返すことになります。当然、極性の高い固定相を使った場合には、極性の高い成分ほどよく吸着して、カラムを抜け出て来るのが遅れます。逆に極性の低い固定相を使った場合には、試料の極性が吸着にあまり効きませんので、他に目立った相互作用がなければ、単純に気化しやすい順、つまり沸点の低い順に出て来る傾向が見られます。つまり、図2(c)の充填式蒸留塔と同じことになるのです。
固定相に液体を使う場合は、HPLCと同じように何かに固定する必要があります。とは言っても、流れて来るのがガスですから、図6のような強力な化学結合までは必要なく、珪藻土などの多孔性の固体に浸み込ませるぐらいで十分です。つまり、担体表面と反応するような特殊な物質を使う必要はなく、極端に言えば、シリコーンオイルのような気化しにくい液体なら何でも利用できるのです。GCではHPLCのように移動相側で条件を変えることが難しい反面、固定相側の選択肢は非常に多く、自分で特別なカラムを作るのも簡単なのです。
GCで特徴的なカラムとして、「キャピラリーカラム」というのがあります。液相カラムの一種で、充填剤を使わず、管の内壁に直にシリコーンオイルなどの液相をコーティングしてあります。材質は
石英ガラス(溶融石英)ですが、外側に保護材としてポリイミドという茶色の樹脂をコートしてあるため、色は茶色。管の内径は0.2mm〜0.5mmと極細で、20m〜50mぐらいの長い管をコイルのようにグルグル巻きにしてありますので、一目でそれとわかります。このカラムの特徴は何と言っても分離性能の高さです。
普通の充填式のカラムの場合、充填剤の隙間をキャリアーガスが流れるのですが、隙間の状態は一様ではなく、様々なルートがあります。図7(a)のように、あるルートは右に左にクネクネと曲がっているでしょうし、あるルートは幅広く直線的かもしれません。こうなると、ルートによってガスが通り抜けるまでの時間にずいぶん差ができます。クロマトグラフィーでは、カラムから出て来るまでの時間(保持時間)の差で成分を分けるのですから、同じ成分は同じ時間にピタッと揃って出て来て欲しいわけですが、これでは同じ成分でありながら、ある幅を持ってダラダラと出て来ることになってしまいます。クロマトグラムで言えば、図の右側に示したようにピークの幅が広がるのです。その点キャピラリーカラムでは、ルートは一本だけで、近道も遠回りもありませんから時間差は生じません。図7(b)のように各成分はピタッと決まった時間に出て来て鋭いピークとなり、他の成分と重ならないので、充填カラムで分離できないような成分もきっちり分離できるようになります。
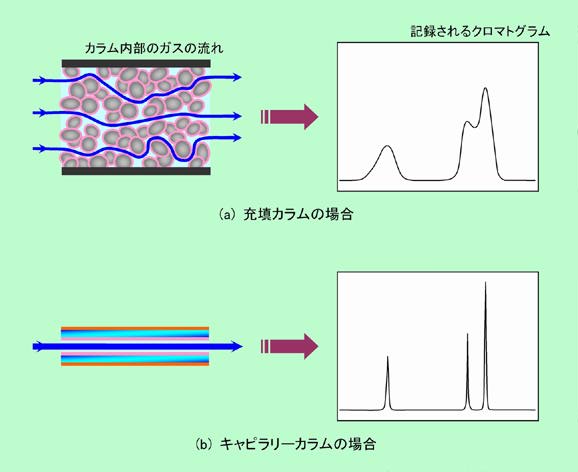
図7 充填カラムとキャピラリーカラム(かなり誇張してます)
GCの検出器としては、熱伝導度を測るものや、試料を燃やして電気伝導を測るものなどが使われます。熱伝導度検出器(Thermal Conductivity Detector = TCD)は、電流を流してフィラメントを加熱しておき、ここにガスを流して温度の変化を検出する装置です。普通にキャリアーガスとして使われるヘリウムは熱伝導度が高い(熱は気体分子が動いて衝突することで運ばれますが、ヘリウムは軽いので動きが速く、熱を伝えるのが速い)ので、他のガスが混じると熱伝導度が下がり、フィラメントの温度が上がるのです(水素だけはヘリウムよりも軽いので、逆になります)。乱暴な言い方をすれば、フィラメントの冷やされ方が変わる、ということですね。この原理からわかるように、TCDはキャリアーガス以外ならどんなガスでも検出できますが、感度はそれほど高いとは言えません。一方、水素炎イオン化検出器(Flame Ionization Detector = FID)は、カラムから出て来たガスに水素と空気を混ぜて火を付け、成分が燃えた時に発生するイオンを検出する装置です。水素炎の上には電圧をかけたキャップがかぶせてあり、イオンが発生した時に流れる電流を捕まえるのです。当然ながら燃えない物は測れないわけですが、分析対象として圧倒的に多いのは有機物ですから、ほとんどの場合、FIDで検出できます。感度もTCDよりも高いので、市販のGCの装置には、たいてい標準で装備されています。
HPLCの場合と同じように、質量分析器を検出器として持っているGCもあります。これらの他にも様々な検出器がありますが、どちらかと言うと特殊な分析用のものが多いので、ここでは省略します。
超臨界流体クロマトグラフィー(SFC)
移動相として、液体や気体の代わりに
超臨界流体を使ったクロマトグラフィーです。
超臨界流体は、温度、圧力を高くすることで気体状態と液体状態の区別がなくなった状態の流体のことで、いろいろな性質が気体と液体の中間の値を示します。密度は気体よりも液体に近く、それだけいろいろな物質を溶かしやすくなる一方で、粘度は普通の液体よりもはるかに低いため、狭い隙間にも簡単に入り込み、気体のようにスムーズに移動できます。というわけで、これをクロマトグラフィーの移動相に利用すると、GCよりもシャープな分離性能で、HPLCよりもスピーディーな分析ができる、ということになります。もっともこれは裏を返せば、GCよりも遅く、HPLCよりも分離が悪い、ということにもなるわけで、要は目的に合った使い分けをすればよいのです。
超臨界流体と言うと、何か特殊な、扱いにくい物のように聞こえるかもしれませんが、よく使われる二酸化炭素の場合、31℃、72気圧を超えると超臨界流体になってしまいます。温度は真夏の気温より低いぐらいですし、圧力も、HPLCのポンプで簡単に作れるレベルですから、設備的には普通のHPLCの装置にちょっと手を加えるだけで事足りるのです。大きな違いと言えば、圧力が下がらないように、出口のところにバルブをつける必要があることぐらいでしょう。そして、出口のバルブを抜けて来ると超臨界二酸化炭素は普通のガス状態になって逃げてしまいますから、後には乾燥した試料が残って簡単に回収できる、というオマケも付きます。というわけでSFCの設備は、分析だけでなく、成分の分離回収にもよく使われるのです(
超臨界流体の話参照)。
ゲル浸透クロマトグラフィー(GPC)、ゲルろ過クロマトグラフィー(GFC)
これは液体クロマトグラフィーの一種ですが、成分を保持するのに図1(c)の原理を使います。装置の構成や外観はHPLCとほとんど変わりませんが、一般的にカラムは長めで30cmぐらいあり、さらにそれを2,3本つなげて使うこともあります(20cm以下の短いカラムもあります)。カラムに詰められている固定相は網目構造をしたゲルで、これには大小様々な大きさの孔が無数に開いています。大きな孔の壁には小さな孔が開いており、その小さな孔の壁にはさらに小さな孔が開く、というふうに、奥へ進むほど孔が小さくなる
フラクタル的な構造になっているのが普通です。ここに試料の分子がやって来ると、小さい分子は小さな孔にも入りますから、ゲルの奥の方まで入り込むことができます。これに対して大きな分子は、あまり奥の方にまでは進むことができません。その結果、寄り道ができない大きな分子ほど速くカラムを通り抜け、たくさん寄り道ができる小さな分子ほど遅れて出て来ることになるのです。この方式のクロマトグラフィーのことを「サイズ排除クロマトグラフィー」と呼びますが、そのうち、移動相に有機溶媒を使うものを「ゲル浸透クロマトグラフィー(Gel Permeation Chromatography = GPC)」、水を使うものを「ゲルろ過クロマトグラフィー(Gel Filtration Chromatography = GFC)」と呼んで区別することもあります。もっとも、「ろ過」というのは大きな物を止めて小さな物を流す操作ですから、この場合は逆ですが・・・・・。
ゲルの材料には、ポリスチレン、アクリルなどのポリマーやセルロース、アガロース(寒天の主成分)などの糖類がよく使われます。また水系のGFC専用ですが、シリカゲルが使われることもあります。GPCもGFCも分子の大きさ、分子量を調べる目的で使われるのですが、GPCが合成ポリマーの分子量分布測定によく用いられるのに対して、GFCはタンパク質などの生体関連分子の分析を中心に活躍しています。
イオンクロマトグラフィー(IC)
これも液体クロマトグラフィーの一種で、固定相にイオン交換樹脂、移動相に「溶離液」と呼ばれる電解質の水溶液を使って、図1(d)の原理でイオンを専門に分離するクロマトグラフィーです。試料がない状態では、溶離液中のイオンの一部が樹脂に捕まって(もちろん「平衡状態」ですから、付いたり離れたりを常に繰り返しながら)一定の状態を保っています。ここに別のイオンを含んだ試料が送り込まれて来ると、試料中のイオンは樹脂を無視して素通りすることはできず、溶離液のイオンと入れ替わって樹脂に捕まったり、また入れ替わって離れたりを繰り返しながら進んで行くことになります。当然、イオンの種類によって樹脂との相性には差があり、樹脂に捕まりやすいイオンほど進行が遅くなりますから、いろいろなイオンが種類ごとに分離されて行くのです。ナトリウムや鉄などの金属イオンや、塩化物イオン、フッ化物イオンなどのハロゲンイオン、硫酸イオン、硝酸イオン等々、たいていのイオンは分析可能で、タンパク質などもpHを適当に調整してイオンにしてやれば分析できます。ただし陽イオン用のイオン交換樹脂と陰イオン用のイオン交換樹脂は全く別のものですから、ICのカラムも、陽イオン用と陰イオン用を別々に用意する必要があります。
イオンクロマトグラフィーの検出器にもいろいろなタイプがありますが、普通は電気伝導度を測定する装置が使われます。ただ、溶離液も電解質ですから、濃い溶離液を使った場合には試料の検出がしづらくなってしまいます。そこで、サプレッサという装置を検出器の前に取り付けて邪魔なイオンを除去する方法もあります。
タンパク質などの場合は、図5(b)のタイプのカラムで、分析ではなく分離精製を目的としてイオンクロマトグラフィーを使うこともあります。この場合は、まず試料をカラムの入り口付近にドカッと吸着させ、次いで別の液を流して吸着したタンパク質を流し出す、という方法が採られます。具体的には、pHを徐々に変えて行って、イオンの状態でいられなくなったものから順に流し出したり(タンパク質はpHによって陽イオンになったり陰イオンになったり、その間ではほとんどイオンにならなかったりします)、塩化ナトリウムなどの水溶液を、薄い溶液から徐々に濃度を上げながら送り込んで、付着力の弱いものから順に置き換えて流し出したりします。
これらの他に、図1(e)でもちょっと触れた、特定の成分とだけ結合するような物質を固定相に植え付けて分離する「アフィニティ クロマトグラフィー(Affinity Chromatography)」や、タンパク質などが水になじみにくい性質を利用して分離する「疎水性(相互作用)クロマトグラフィー(Hydrophobic Interaction Chromatography)」などがありますが、詳しい説明はここでは省略します。
クロマトグラフィーで成分が分離される様子
「固定相に捕まりやすいかどうかでカラムを通り抜ける速度が変わり、それによって成分が分離される」、ということをこれまで何回も書いて来ました。図4にもその様子が大雑把に示されてはいますが、これをもう少し厳密に見てみることにします。その際、クロマトグラフィーのカラムは本来は図4(b)のような連続的なものなのですが、これでは計算しにくいので、図4(a)のように小部屋に区切られていると考えます。この各部屋を「段」と呼ぶことにしましょう。各段で、成分は移動相と固定相にしっかりと分配され(つまり完全に平衡状態になり)、その後、移動相がそれぞれ隣の段に移って行くと考えます。この各段階をStepと呼ぶことにします。例えばStep10では、移動相の先頭は10番目の段に来ており、成分はそれよりも遅れて途中の段に分布している、というわけです。
このような段に分かれたカラムの中を、成分がどのように移動していくかを示したのが図8です。移動相と固定相への分配比率を3:7と仮定し、仕込んだ全量を1として移動相中の成分量を計算しました(固定相中の量は示していませんが、7/3倍になるだけで、パターンは全く同じです)。
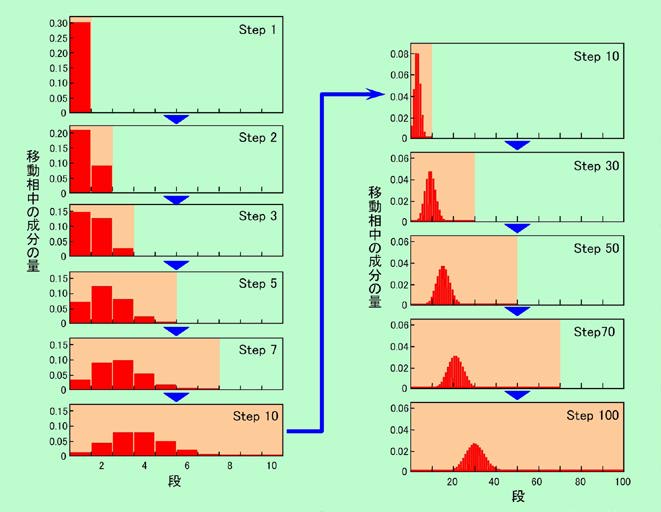
図8 カラム中を成分が移動する様子
左の列はStep1からStep10までを示しています。Step1では全量が1段目にあるので、その中の3割が移動相に来ています。これが、Stepが進むにつれて右に移動して行き、最後のStep10では、移動相の先頭は10段目に、そして成分の方はかなり遅れて、3段目、4段目あたりを頂点に、裾の広い分布になっています。ここからさらに進んだ場合を示したのが右側の列です(一番上は左下と同じStep10ですが、横軸スケールが変わっています)。Stepが進むにつれて成分は右に移動して行くのですが、先頭からの遅れはどんどん大きくなり、また左右に大きく広がって来ることがわかります。カラムの中を長く移動するほど分布がシャープになるかと思いきや、実は逆に広がっているのです。
ところが、移動相が進んだ距離を基準にして見直してみると、様子がずいぶん変わって来ます。図9は、移動相の進んだ距離が横幅いっぱいになるように横軸スケールを変えて、成分の分布を書き直したものですが(ピークの高さも揃えています)、Stepが進むほどシャープな分布になり、左右対称のきれいな形(正規分布に近い形)になって来ることがわかります。Step数が少ない時は成分の広がりは確かに少ないのですが、移動相自体もそれほど進んでいませんから、移動相が進んだ距離を基準にした時の広がり幅はむしろ大きくなってしまうのです。
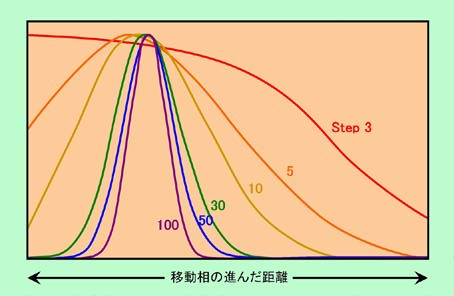
図9 移動相が動いた距離を基準にして成分の分布を見直してみると・・・・
図9は見方を変えれば、「段数の違うカラムを比較したもの」と考えることもできます。例えば赤線で示されたStep3のカーブは、「長いカラムの最初の3段分だけ移動相が進んだ時の成分の分布を表したもの」とも言えますし、「3段しかないカラムで移動相の先頭が出口に到達した時の成分の分布を表したもの」とも言えるのです。こう考えると、長さが同じならば、「段数が多いカラムの方が成分の分布は狭く、形も左右対称できれい」と言えることがわかります。実はこの特徴は、クロマトグラフィーの本来の目的である成分の分離には直接に影響します。その様子を示したのが図10です。この図では、成分Aは図8と同じように移動相と固定相に3:7に分配されるのに対して、成分Bは固定相に保持されにくく、分配比率は6:4であると仮定して計算しています。図10(a)は100段のカラムの中で2種類の成分がどのように動いて行くかを示したもの、図10(b)は段数の違うカラムで、移動相の先頭が出口に到達した時に、2種類の成分がどのように分布しているかを示したものですが、これらは横軸の幅を変えて同じデータを書き直したに過ぎません(図8―図9の関係と全く同じ)。
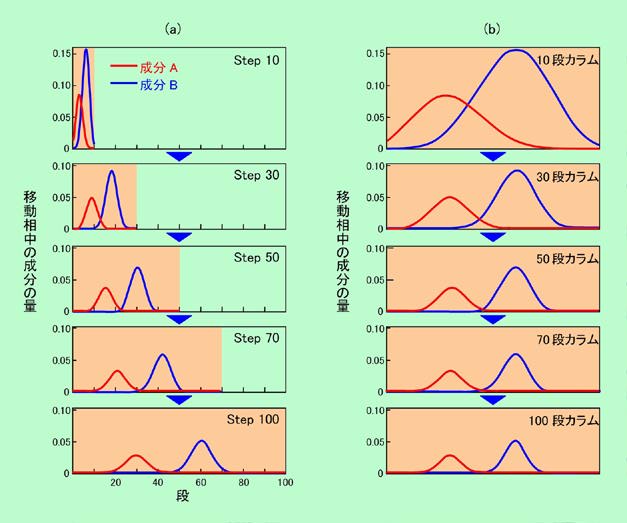
図10 2種類の成分の分離
図10(a)を見ると、固定相に保持されやすい成分Aの方が移動が遅く、2つの成分がしだいに分離して来ることがわかります。Stepが進むほど2成分の差が広がり、分離がよくなるのです。これは言い換えると、図10(b)に示されているように、段数の少ないカラムでは成分の分離が十分にはできない、ということでもあります。この例で言えば、成分Aと成分Bを完全に分離するには、少なくとも70段ぐらいのカラムが必要、ということですね。つまり段の数というのは、カラムの分離性能そのものなのです。
実際のカラムでも、分離性能を表すのに、この「段数」の考え方を使います。現実のカラムは段に分かれているわけではありませんが、「もし段に分かれていたとしたら何段のカラムに相当するか」を考えるのです。これが「理論段数」です。もちろん、理論段数が大きいほど成分はゆっくり移動して明瞭に分離されますし、成分の分布、つまり検出器で検出されるピークもシャープできれいな形になります。逆に、成分が出て来るまでの時間とピークの形から理論段数を計算することも可能で、市販のカラムのカタログなどに載っている理論段数の値は、実際の試料の分析結果から算出したものです。もちろん測定条件によって性能は変わって来ますから、理論段数は絶対的な数値ではありませんが・・・・。
図8〜10に示した例と違って、実際のクロマトグラフィーで使われるカラムの理論段数は非常に大きく、数千から数万、ものによっては数十万にもなります。精密な分析をするには、これくらいの段数が必要なのです。なお、一つ付け加えておくと、図2(b)の棚段式蒸留塔のような本当に段に分かれている装置の場合でも、構造上の段の数と理論段数とは必ずしも一致はしません。理論段数の考え方では、それぞれの段で完全な平衡状態ができることや、その段の移動相が100%余すところなく次の段に移動すること、などが仮定されていますが、実際にはそうは行かないからです。「理論段数」はあくまでも「理論上の」段数なのです。
ところで、図8〜10に示した成分の分布は、あくまでもカラムの内部での分布です。クロマトグラフの検出器はカラムの外にあるわけですから、実際にピークとして観測されるのはカラムから出た後の成分量のはずですね。実は、カラムから出た後の成分分布の形は、カラム内部とはちょっと変わるのです。
図11は、固定相と移動相の分配比率が6:4の成分を40段のカラムに流した時に、カラムの出口でピークの形がどのように変わるかを図示したものです。ここで示しているのは移動相中の成分量で、左側の薄い赤色に着色してある部分がカラムの内部です。
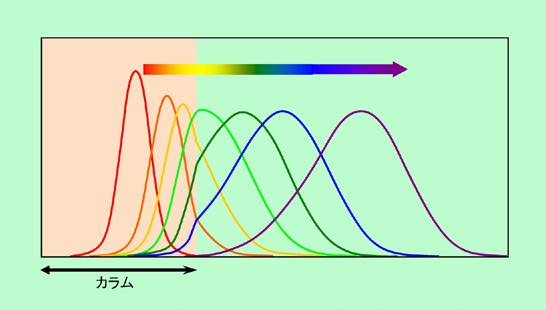
図11 カラムの出口でピークの形が変わる
図からわかるように、カラムの出口のところで、成分の量を示す曲線がカクッと折れ曲がります。そして成分がカラムから出始める直前には赤線で示したような形であったのが、ほぼ全量がカラムを抜け出た時点では、紫線のように幅が広がり、左側(つまり後ろ側)に尾を引いたような形に変わってしまうのです。
なぜこのようなことが起こるのかを示したのが図12です。これはカラムの最後の2段を示したもので、青い棒が固定相に保持された成分量、赤やピンクの棒が移動相中の成分量を表します。
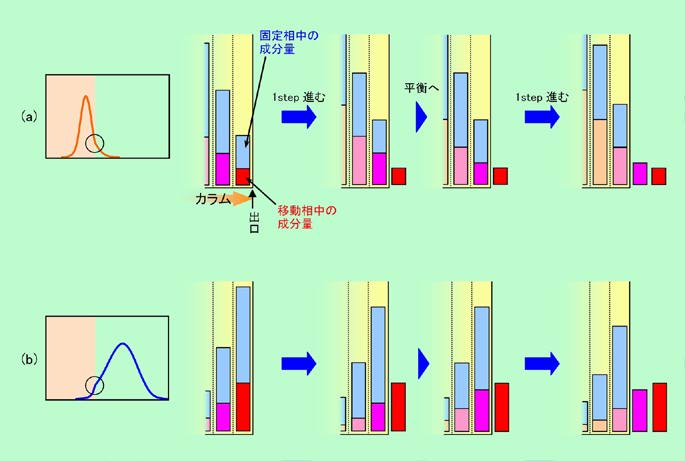
図12 カラムの出口付近での成分量の変化
まずピーク前半の、成分量が徐々に増えて行く部分がカラムを抜け出して来る場合を見てみましょう。図12(a)の最初の状態では、固定相と移動相への分配比率はどの段でも一定です(平衡状態)。ここから1Step進むと、最終段の赤棒の部分がカラムの外へ出て、一つ前のピンク棒の部分が最終段に移って来ます。すると、ピンク棒は赤棒よりも長いですから、最終段の固定相と移動相の平衡状態が崩れますので、移動相中の成分の一部が固定相へ移り、元の平衡状態に戻ります。つまりピンクの棒が少し短くなるわけです。この短くなったピンクの棒が次のStepでカラムから出て来ることになります。このように、後から出て来る成分の量は、カラム中にいた時と比べて常に少ない状態になりますから、ピーク前半の傾斜は、カラムを出ると緩やかになってしまうのです。
ピーク後半の下りの部分が出口にさしかかると、図12(b)のように逆のパターンになります。赤棒より短いピンク棒が最終段に移って来ると、平衡状態に戻すために、固定相から移動相に成分が移動し、ピンク棒は元より長くなります。これがカラムから出て来るわけですから、後から出て来る成分の量は、前半とは逆にカラム中にいた時と比べて常に多い状態になります。その結果、ピーク後半の下りの傾斜も緩くなり、前も後ろも緩やかな、横に広がった形になってしまうのです。
この現象は、次のように簡単に考えることもできます。図8などで示しているように、カラムの中を移動している間に成分の分布はしだいに広がって来ます。一方、カラムを出た後は、もう分布が広がることはありません。というわけで、早い段階でカラムを抜け出た部分はあまり形が変わらないのに対して、後ろの方に行くほどカラム内に長く残るために幅が広がって来ます。その結果、後ろの方ほど横に引き伸ばされた、後ろ髪を引かれたような状態になってしまうのです。
このような形の変化は、段数が多い分析用のカラムなどではあまり目立ちません。ピークが理想の形から崩れる原因は、むしろ成分とカラムとの特殊な相互作用やカラムの汚れなど、別のところにある場合がほとんどです。このあたりの内容まで踏み込むとまた込み入った話になりますので、ここは専門の解説書等に任せるとして、本項では理想的なケースにとどめておくことにします。
雑科学ホーム
hr-inoueホーム